Ruddlesden-Popper结构杂化非常规铁电体的研究进展
张碧辉, 刘小强, 陈湘明
无机材料学报
2025, 40 ( 6):
587-608.
DOI:10.15541/jim20240521
杂化非常规铁电性(Hybrid Improper Ferroelectricity, HIF)指的是在含钙钛矿结构单元的化合物中, 通过阴离子八面体面内旋转和面外倾侧耦合而产生的二阶铁电序。HIF有望在强磁电耦合多铁性材料中获得重要应用, 并极大地拓展铁电体物理学的内涵和外延。本文总结了Ruddlesden-Popper(R-P)结构HIF的实验研究进展, 建立了双层R-P结构铁电体的居里温度(TC)和许容因子(τ)之间的线性关系, 并阐述其HIF物理起源。基于HIF的内禀电控磁性, 在双层R-P铁氧体中观察到室温极性相和弱铁磁相共存, 这一发现具有重要的科学意义。此外, 在A位离子有序三层R-P氧化物中报道的铁电性显著拓宽了HIF的研究广度和深度。尽管R-P结构的HIF的研究已取得显著进展, 但在新材料体系和单相多铁性材料探索方面仍需进一步努力。
View image in article
图9
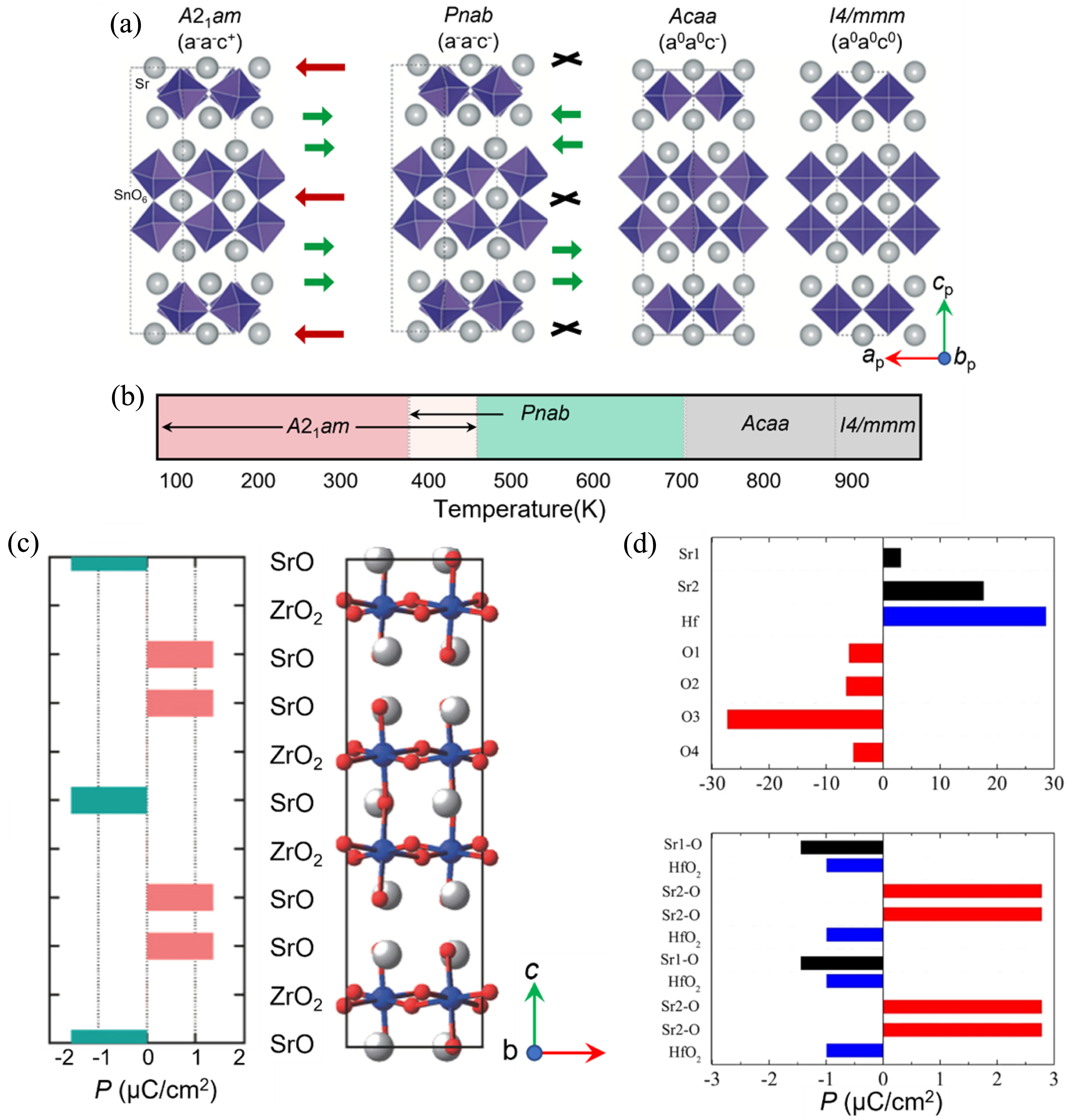
(a)根据空间群对称性分析得到Sr3Sn2O7随温度变化的四个相结构[118]; (b)研究建立的Sr3Sn2O7的相图[118]; (c)根据300 K条件下的NPD精修结果计算的晶体结构分层极化(左图), [010]面的晶体结构示意图(右图), 其中Sr、Zr和O原子分别为灰色、蓝色和红色[119]; (d) Sr3Hf2O7中原子和分层极化对宏观极化的贡献, Sr1−O表示钙钛矿层之间的SrO层, Sr2−O表示岩盐层和钙钛矿层之间的SrO层[120]
正文中引用本图/表的段落
为了拓展HIF的研究, Yoshida等[119]发现Sr3Zr2O7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A21am。结合SHG信号、同步辐射XRD和NPD结果, 确定Sr3Zr2O7属于极性A21am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如图9(c)所示。Sr3Zr2O7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[119]。SHG信号表明Sr3Zr2O7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A21am结构。Sr3Zr2O7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm。其铁电极化强度与Ca3Ti2O7陶瓷 (0.6 μC/cm2)[43]和Sr3Sn2O7陶瓷(0.25 μC/cm2)[117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级。虽然在(Sr,Ba)3Zr2O7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[86]。Sr3M2O7化合物普遍经历复杂的相变过程, I4/mmm到A21am的相变过程中经历了两次二级相变(图9(a, b))[118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解。这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[59-60]。基于此, 本课题组[120]通过DFT计算揭示了Sr3Hf2O7的HIF和铁电翻转路径, 发现从Sr3Hf2O7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr3Hf2O7相对较低的理论极化强度是由HfO2层的负贡献引起, 如图9(d)所示。
尽管理论上已经预测并成功合成了许多具有单层R-P结构的非中心对称结构氧化物, 并对部分单层R-P氧化物的压电特性进行了表征, 但其可翻转的铁电极化鲜有报道[139-140,148]。本课题组通过放电等离子烧结工艺成功制备了致密的Li2La2Ti3O10陶瓷, 并利用PUND法在室温下观测到了其饱和电滞回线, 测得剩余极化强度为0.25 μC/cm2, 矫顽场为260 kV/cm[149], 如图16(a)所示。结合NPD, 同步辐射XRD和选区电子衍射(SAED)确定Li2La2Ti3O10陶瓷具有极性P21ab空间群的三层R-P结构[149]。此外, DFT计算结果显示Pc、P21ab、P21am(No. 26)、A21am和P21an空间群与Li2La2Ti3O10结构精修的拟合优度χ2分别为6.15、4.82、5.12、4.95、5.43, 加权图形剩余方差因子Rwp分别为9.19%、8.16%、8.41%、8.30%、8.66%, 进一步验证了Li2La2Ti3O10氧化物形成能最低的空间群为P21ab(图16(b))。这项工作首次在实验上证实了A位阳离子有序的三层R-P结构的Li2La2Ti3O10陶瓷在室温下具有铁电性, 极大地扩展了HIF的研究领域[149]。随后, 本课题组基于NPD数据精修确定Li2Nd2Ti3O10陶瓷在室温下属于极性P21ab相[150]。PUND法测得其饱和室温电滞回线, 具有约0.4 μC/cm2的剩余极化强度和150 kV/cm的矫顽场, 如图16(c)所示。Li2Nd2Ti3O10陶瓷的铁电性同样起源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的协同作用[150]。与Li2La2Ti3O10陶瓷相比, Li2Nd2Ti3O10陶瓷表现出更高的铁电极化强度和较低的矫顽场, 这可能是源于Nd3+离子半径小于La3+导致更显著的氧八面体畸变(图16(d)), 进而获得了更优异的铁电性能。而较低的矫顽场则与Li2La2Ti3O10陶瓷含有少量的第二相有关。Li2La2Ti3O10陶瓷中的铁电性源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的耦合, 其中A位阳离子有序是奇数层R-P结构材料获得极性相的关键因素[149], 其晶体结构如图16(e)所示。图16(f)展示了R-P层状结构中各层极化对宏观极化的贡献, 宏观极化主要源于TiO6氧八面体畸变, 这与双层R-P氧化物的铁电起源完全不同; 双层R-P氧化物的宏观极化是由A位阳离子的未抵消反铁畸变位移引起。A位离子有序三层R-P结构陶瓷的实验工作为进一步拓宽HIF的研究宽度与广度提供了新的见解, 且研究思路可以拓展到其余层状钙钛矿氧化物[149-150]。
为了拓展HIF的研究, Yoshida等[ 119]发现Sr 3Zr 2O 7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A2 1am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr 3Zr 2O 7属于极性A2 1am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如 图9(c)所示.Sr 3Zr 2O 7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[ 119].SHG信号表明Sr 3Zr 2O 7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A2 1am结构.Sr 3Zr 2O 7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca 3Ti 2O 7陶瓷 (0.6 μC/cm2)[ 43]和Sr 3Sn 2O 7陶瓷(0.25 μC/cm2)[ 117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba) 3Zr 2O 7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[ 86].Sr 3M 2O 7化合物普遍经历复杂的相变过程, I4/mmm到A2 1am的相变过程中经历了两次二级相变( 图9(a, b))[ 118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[ 59- 60].基于此, 本课题组[ 120]通过DFT计算揭示了Sr 3Hf 2O 7的HIF和铁电翻转路径, 发现从Sr 3Hf 2O 7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr 3Hf 2O 7相对较低的理论极化强度是由HfO 2层的负贡献引起, 如 图9(d)所示. ...
为了拓展HIF的研究, Yoshida等[ 119]发现Sr 3Zr 2O 7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A2 1am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr 3Zr 2O 7属于极性A2 1am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如 图9(c)所示.Sr 3Zr 2O 7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[ 119].SHG信号表明Sr 3Zr 2O 7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A2 1am结构.Sr 3Zr 2O 7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca 3Ti 2O 7陶瓷 (0.6 μC/cm2)[ 43]和Sr 3Sn 2O 7陶瓷(0.25 μC/cm2)[ 117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba) 3Zr 2O 7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[ 86].Sr 3M 2O 7化合物普遍经历复杂的相变过程, I4/mmm到A2 1am的相变过程中经历了两次二级相变( 图9(a, b))[ 118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[ 59- 60].基于此, 本课题组[ 120]通过DFT计算揭示了Sr 3Hf 2O 7的HIF和铁电翻转路径, 发现从Sr 3Hf 2O 7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr 3Hf 2O 7相对较低的理论极化强度是由HfO 2层的负贡献引起, 如 图9(d)所示. ...
为了拓展HIF的研究, Yoshida等[ 119]发现Sr 3Zr 2O 7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A2 1am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr 3Zr 2O 7属于极性A2 1am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如 图9(c)所示.Sr 3Zr 2O 7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[ 119].SHG信号表明Sr 3Zr 2O 7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A2 1am结构.Sr 3Zr 2O 7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca 3Ti 2O 7陶瓷 (0.6 μC/cm2)[ 43]和Sr 3Sn 2O 7陶瓷(0.25 μC/cm2)[ 117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba) 3Zr 2O 7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[ 86].Sr 3M 2O 7化合物普遍经历复杂的相变过程, I4/mmm到A2 1am的相变过程中经历了两次二级相变( 图9(a, b))[ 118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[ 59- 60].基于此, 本课题组[ 120]通过DFT计算揭示了Sr 3Hf 2O 7的HIF和铁电翻转路径, 发现从Sr 3Hf 2O 7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr 3Hf 2O 7相对较低的理论极化强度是由HfO 2层的负贡献引起, 如 图9(d)所示. ...
为了拓展HIF的研究, Yoshida等[ 119]发现Sr 3Zr 2O 7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A2 1am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr 3Zr 2O 7属于极性A2 1am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如 图9(c)所示.Sr 3Zr 2O 7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[ 119].SHG信号表明Sr 3Zr 2O 7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A2 1am结构.Sr 3Zr 2O 7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca 3Ti 2O 7陶瓷 (0.6 μC/cm2)[ 43]和Sr 3Sn 2O 7陶瓷(0.25 μC/cm2)[ 117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba) 3Zr 2O 7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[ 86].Sr 3M 2O 7化合物普遍经历复杂的相变过程, I4/mmm到A2 1am的相变过程中经历了两次二级相变( 图9(a, b))[ 118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[ 59- 60].基于此, 本课题组[ 120]通过DFT计算揭示了Sr 3Hf 2O 7的HIF和铁电翻转路径, 发现从Sr 3Hf 2O 7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr 3Hf 2O 7相对较低的理论极化强度是由HfO 2层的负贡献引起, 如 图9(d)所示. ...
为了拓展HIF的研究, Yoshida等[ 119]发现Sr 3Zr 2O 7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A2 1am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr 3Zr 2O 7属于极性A2 1am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如 图9(c)所示.Sr 3Zr 2O 7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[ 119].SHG信号表明Sr 3Zr 2O 7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A2 1am结构.Sr 3Zr 2O 7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca 3Ti 2O 7陶瓷 (0.6 μC/cm2)[ 43]和Sr 3Sn 2O 7陶瓷(0.25 μC/cm2)[ 117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba) 3Zr 2O 7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[ 86].Sr 3M 2O 7化合物普遍经历复杂的相变过程, I4/mmm到A2 1am的相变过程中经历了两次二级相变( 图9(a, b))[ 118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[ 59- 60].基于此, 本课题组[ 120]通过DFT计算揭示了Sr 3Hf 2O 7的HIF和铁电翻转路径, 发现从Sr 3Hf 2O 7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr 3Hf 2O 7相对较低的理论极化强度是由HfO 2层的负贡献引起, 如 图9(d)所示. ...
为了拓展HIF的研究, Yoshida等[ 119]发现Sr 3Zr 2O 7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A2 1am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr 3Zr 2O 7属于极性A2 1am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如 图9(c)所示.Sr 3Zr 2O 7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[ 119].SHG信号表明Sr 3Zr 2O 7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A2 1am结构.Sr 3Zr 2O 7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca 3Ti 2O 7陶瓷 (0.6 μC/cm2)[ 43]和Sr 3Sn 2O 7陶瓷(0.25 μC/cm2)[ 117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba) 3Zr 2O 7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[ 86].Sr 3M 2O 7化合物普遍经历复杂的相变过程, I4/mmm到A2 1am的相变过程中经历了两次二级相变( 图9(a, b))[ 118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[ 59- 60].基于此, 本课题组[ 120]通过DFT计算揭示了Sr 3Hf 2O 7的HIF和铁电翻转路径, 发现从Sr 3Hf 2O 7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr 3Hf 2O 7相对较低的理论极化强度是由HfO 2层的负贡献引起, 如 图9(d)所示. ...
为了拓展HIF的研究, Yoshida等[ 119]发现Sr 3Zr 2O 7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A2 1am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr 3Zr 2O 7属于极性A2 1am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如 图9(c)所示.Sr 3Zr 2O 7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[ 119].SHG信号表明Sr 3Zr 2O 7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A2 1am结构.Sr 3Zr 2O 7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca 3Ti 2O 7陶瓷 (0.6 μC/cm2)[ 43]和Sr 3Sn 2O 7陶瓷(0.25 μC/cm2)[ 117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba) 3Zr 2O 7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[ 86].Sr 3M 2O 7化合物普遍经历复杂的相变过程, I4/mmm到A2 1am的相变过程中经历了两次二级相变( 图9(a, b))[ 118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[ 59- 60].基于此, 本课题组[ 120]通过DFT计算揭示了Sr 3Hf 2O 7的HIF和铁电翻转路径, 发现从Sr 3Hf 2O 7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr 3Hf 2O 7相对较低的理论极化强度是由HfO 2层的负贡献引起, 如 图9(d)所示. ...
为了拓展HIF的研究, Yoshida等[ 119]发现Sr 3Zr 2O 7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A2 1am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr 3Zr 2O 7属于极性A2 1am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如 图9(c)所示.Sr 3Zr 2O 7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[ 119].SHG信号表明Sr 3Zr 2O 7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A2 1am结构.Sr 3Zr 2O 7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca 3Ti 2O 7陶瓷 (0.6 μC/cm2)[ 43]和Sr 3Sn 2O 7陶瓷(0.25 μC/cm2)[ 117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba) 3Zr 2O 7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[ 86].Sr 3M 2O 7化合物普遍经历复杂的相变过程, I4/mmm到A2 1am的相变过程中经历了两次二级相变( 图9(a, b))[ 118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[ 59- 60].基于此, 本课题组[ 120]通过DFT计算揭示了Sr 3Hf 2O 7的HIF和铁电翻转路径, 发现从Sr 3Hf 2O 7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr 3Hf 2O 7相对较低的理论极化强度是由HfO 2层的负贡献引起, 如 图9(d)所示. ... Distortion modes and ferroelectric properties in hybrid improper ferroelectric Sr3(Sn, Zr)2O7 ceramics 2 2022 ... Sr3Sn2O7氧化物具有双层R-P结构, 其晶格参数为a=5.710 ?、b=5.736 ?和c=20.688 ?[114].尽管早期中子粉末衍射(NPD)实验显示, Sr3Sn2O7属于正交的Cmcm空间群[115], 但可能通过复杂晶格畸变的冻结而诱发HIF[15,26,49,116].2017年, Wang等[117]在双层R-P结构的Sr3Sn2O7陶瓷中首次观测到HIF, 利用XRD精修确定了Sr3Sn2O7在室温下属于非中心对称极性空间群A21am, 并通过PUND法获得了典型的室温电滞回线, 其剩余极化强度和矫顽场分别为0.1 μC/cm2和200 kV/cm.随后, Xu等[112]报道称Sr3Sn2O7单晶沿[100]的矫顽场最低可达39 kV/cm, 这是目前已知HIF体系中最小的室温矫顽场, 与IP-PFM观测到带电DWs和中性DWs有关.结合Yoshida等[118]和本课题组的理论计算与实验数据, Sr3Sn2O7的相变历程为I4/mmm→Acaa→Pnab→A21am.Yoshida和本课题组总结了双层R-P材料的TC与τ呈现线性负相关的关系, 发现TC随着Ca2+含量的增加而线性增加, 该线性关系适用于所有双层R-P结构A3B2O7的HIF[118].本课题组[84]通过高能球磨提高了煅烧粉末的烧结活性, 在高质量的致密Sr3?xBaxSn2O7陶瓷中获得了0.26 μC/cm2的铁电极化强度和260 kV/cm的矫顽场, 与Sr3Sn2O7陶瓷相比, 铁电性能有所改善, 这归因于τ的降低.Sr3Sn2O7陶瓷的铁电极化强度为0.08 μC/cm2, 矫顽场为335 kV/cm.由于Ca3Sn2O7具有较大的理论极化强度, 在Sr3Sn2O7陶瓷的A位引入Ca2+可以有效改善铁电性, 且TC随着Ca2+含量增加而升高[85].在这些样品中, Sr3Sn2O7单晶表现出最出色的铁电性能, 沿[100]面测得的铁电极化强度为1.1 μC/cm2, 矫顽场为39 kV/cm[112].典型双层R-P结构Sr基氧化物的铁电性能对比如图8所示, 基于Sr3Sn2O7陶瓷进行A位置换得到的(Sr,Ca)3Sn2O7陶瓷的矫顽场从335 kV/cm降低至200 kV/cm左右, 铁电极化强度与Sr3Sn2O7陶瓷相当[85], 而A位置换的(Sr,Ba)3Sn2O7陶瓷相对Sr3Sn2O7陶瓷而言获得了较高的铁电极化强度[84].基于Sr3Zr2O7陶瓷的A位置换提高了(Sr,Ba)3Zr2O7陶瓷的铁电极化强度, 但矫顽场显著增高, 而B位置换的Sr3(Sn,Zr)2O7陶瓷的铁电性能与Sr3Zr2O7陶瓷较为接近.
为了拓展HIF的研究, Yoshida等[ 119]发现Sr 3Zr 2O 7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A2 1am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr 3Zr 2O 7属于极性A2 1am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如 图9(c)所示.Sr 3Zr 2O 7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[ 119].SHG信号表明Sr 3Zr 2O 7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A2 1am结构.Sr 3Zr 2O 7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca 3Ti 2O 7陶瓷 (0.6 μC/cm2)[ 43]和Sr 3Sn 2O 7陶瓷(0.25 μC/cm2)[ 117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba) 3Zr 2O 7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[ 86].Sr 3M 2O 7化合物普遍经历复杂的相变过程, I4/mmm到A2 1am的相变过程中经历了两次二级相变( 图9(a, b))[ 118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[ 59- 60].基于此, 本课题组[ 120]通过DFT计算揭示了Sr 3Hf 2O 7的HIF和铁电翻转路径, 发现从Sr 3Hf 2O 7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr 3Hf 2O 7相对较低的理论极化强度是由HfO 2层的负贡献引起, 如 图9(d)所示. ...
为了拓展HIF的研究, Yoshida等[ 119]发现Sr 3Zr 2O 7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A2 1am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr 3Zr 2O 7属于极性A2 1am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如 图9(c)所示.Sr 3Zr 2O 7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[ 119].SHG信号表明Sr 3Zr 2O 7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A2 1am结构.Sr 3Zr 2O 7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca 3Ti 2O 7陶瓷 (0.6 μC/cm2)[ 43]和Sr 3Sn 2O 7陶瓷(0.25 μC/cm2)[ 117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba) 3Zr 2O 7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[ 86].Sr 3M 2O 7化合物普遍经历复杂的相变过程, I4/mmm到A2 1am的相变过程中经历了两次二级相变( 图9(a, b))[ 118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[ 59- 60].基于此, 本课题组[ 120]通过DFT计算揭示了Sr 3Hf 2O 7的HIF和铁电翻转路径, 发现从Sr 3Hf 2O 7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr 3Hf 2O 7相对较低的理论极化强度是由HfO 2层的负贡献引起, 如 图9(d)所示. ... Strong reddish-orange light emission from stress-activated Srn+1SnnO3n+1:Sm3+ (n= ?1, 2, ∞) with perovskite-related structures 1 2012 ... Sr3Sn2O7氧化物具有双层R-P结构, 其晶格参数为a=5.710 ?、b=5.736 ?和c=20.688 ?[114].尽管早期中子粉末衍射(NPD)实验显示, Sr3Sn2O7属于正交的Cmcm空间群[115], 但可能通过复杂晶格畸变的冻结而诱发HIF[15,26,49,116].2017年, Wang等[117]在双层R-P结构的Sr3Sn2O7陶瓷中首次观测到HIF, 利用XRD精修确定了Sr3Sn2O7在室温下属于非中心对称极性空间群A21am, 并通过PUND法获得了典型的室温电滞回线, 其剩余极化强度和矫顽场分别为0.1 μC/cm2和200 kV/cm.随后, Xu等[112]报道称Sr3Sn2O7单晶沿[100]的矫顽场最低可达39 kV/cm, 这是目前已知HIF体系中最小的室温矫顽场, 与IP-PFM观测到带电DWs和中性DWs有关.结合Yoshida等[118]和本课题组的理论计算与实验数据, Sr3Sn2O7的相变历程为I4/mmm→Acaa→Pnab→A21am.Yoshida和本课题组总结了双层R-P材料的TC与τ呈现线性负相关的关系, 发现TC随着Ca2+含量的增加而线性增加, 该线性关系适用于所有双层R-P结构A3B2O7的HIF[118].本课题组[84]通过高能球磨提高了煅烧粉末的烧结活性, 在高质量的致密Sr3?xBaxSn2O7陶瓷中获得了0.26 μC/cm2的铁电极化强度和260 kV/cm的矫顽场, 与Sr3Sn2O7陶瓷相比, 铁电性能有所改善, 这归因于τ的降低.Sr3Sn2O7陶瓷的铁电极化强度为0.08 μC/cm2, 矫顽场为335 kV/cm.由于Ca3Sn2O7具有较大的理论极化强度, 在Sr3Sn2O7陶瓷的A位引入Ca2+可以有效改善铁电性, 且TC随着Ca2+含量增加而升高[85].在这些样品中, Sr3Sn2O7单晶表现出最出色的铁电性能, 沿[100]面测得的铁电极化强度为1.1 μC/cm2, 矫顽场为39 kV/cm[112].典型双层R-P结构Sr基氧化物的铁电性能对比如图8所示, 基于Sr3Sn2O7陶瓷进行A位置换得到的(Sr,Ca)3Sn2O7陶瓷的矫顽场从335 kV/cm降低至200 kV/cm左右, 铁电极化强度与Sr3Sn2O7陶瓷相当[85], 而A位置换的(Sr,Ba)3Sn2O7陶瓷相对Sr3Sn2O7陶瓷而言获得了较高的铁电极化强度[84].基于Sr3Zr2O7陶瓷的A位置换提高了(Sr,Ba)3Zr2O7陶瓷的铁电极化强度, 但矫顽场显著增高, 而B位置换的Sr3(Sn,Zr)2O7陶瓷的铁电性能与Sr3Zr2O7陶瓷较为接近. ... Structure of the n=2 and n=∞ member of the Ruddlesden-Popper series, Srn+1SnnO3n+1 1 2000 ... Sr3Sn2O7氧化物具有双层R-P结构, 其晶格参数为a=5.710 ?、b=5.736 ?和c=20.688 ?[114].尽管早期中子粉末衍射(NPD)实验显示, Sr3Sn2O7属于正交的Cmcm空间群[115], 但可能通过复杂晶格畸变的冻结而诱发HIF[15,26,49,116].2017年, Wang等[117]在双层R-P结构的Sr3Sn2O7陶瓷中首次观测到HIF, 利用XRD精修确定了Sr3Sn2O7在室温下属于非中心对称极性空间群A21am, 并通过PUND法获得了典型的室温电滞回线, 其剩余极化强度和矫顽场分别为0.1 μC/cm2和200 kV/cm.随后, Xu等[112]报道称Sr3Sn2O7单晶沿[100]的矫顽场最低可达39 kV/cm, 这是目前已知HIF体系中最小的室温矫顽场, 与IP-PFM观测到带电DWs和中性DWs有关.结合Yoshida等[118]和本课题组的理论计算与实验数据, Sr3Sn2O7的相变历程为I4/mmm→Acaa→Pnab→A21am.Yoshida和本课题组总结了双层R-P材料的TC与τ呈现线性负相关的关系, 发现TC随着Ca2+含量的增加而线性增加, 该线性关系适用于所有双层R-P结构A3B2O7的HIF[118].本课题组[84]通过高能球磨提高了煅烧粉末的烧结活性, 在高质量的致密Sr3?xBaxSn2O7陶瓷中获得了0.26 μC/cm2的铁电极化强度和260 kV/cm的矫顽场, 与Sr3Sn2O7陶瓷相比, 铁电性能有所改善, 这归因于τ的降低.Sr3Sn2O7陶瓷的铁电极化强度为0.08 μC/cm2, 矫顽场为335 kV/cm.由于Ca3Sn2O7具有较大的理论极化强度, 在Sr3Sn2O7陶瓷的A位引入Ca2+可以有效改善铁电性, 且TC随着Ca2+含量增加而升高[85].在这些样品中, Sr3Sn2O7单晶表现出最出色的铁电性能, 沿[100]面测得的铁电极化强度为1.1 μC/cm2, 矫顽场为39 kV/cm[112].典型双层R-P结构Sr基氧化物的铁电性能对比如图8所示, 基于Sr3Sn2O7陶瓷进行A位置换得到的(Sr,Ca)3Sn2O7陶瓷的矫顽场从335 kV/cm降低至200 kV/cm左右, 铁电极化强度与Sr3Sn2O7陶瓷相当[85], 而A位置换的(Sr,Ba)3Sn2O7陶瓷相对Sr3Sn2O7陶瓷而言获得了较高的铁电极化强度[84].基于Sr3Zr2O7陶瓷的A位置换提高了(Sr,Ba)3Zr2O7陶瓷的铁电极化强度, 但矫顽场显著增高, 而B位置换的Sr3(Sn,Zr)2O7陶瓷的铁电性能与Sr3Zr2O7陶瓷较为接近. ... Polar octahedral rotations: a path to new multifunctional materials 1 2012 ... Sr3Sn2O7氧化物具有双层R-P结构, 其晶格参数为a=5.710 ?、b=5.736 ?和c=20.688 ?[114].尽管早期中子粉末衍射(NPD)实验显示, Sr3Sn2O7属于正交的Cmcm空间群[115], 但可能通过复杂晶格畸变的冻结而诱发HIF[15,26,49,116].2017年, Wang等[117]在双层R-P结构的Sr3Sn2O7陶瓷中首次观测到HIF, 利用XRD精修确定了Sr3Sn2O7在室温下属于非中心对称极性空间群A21am, 并通过PUND法获得了典型的室温电滞回线, 其剩余极化强度和矫顽场分别为0.1 μC/cm2和200 kV/cm.随后, Xu等[112]报道称Sr3Sn2O7单晶沿[100]的矫顽场最低可达39 kV/cm, 这是目前已知HIF体系中最小的室温矫顽场, 与IP-PFM观测到带电DWs和中性DWs有关.结合Yoshida等[118]和本课题组的理论计算与实验数据, Sr3Sn2O7的相变历程为I4/mmm→Acaa→Pnab→A21am.Yoshida和本课题组总结了双层R-P材料的TC与τ呈现线性负相关的关系, 发现TC随着Ca2+含量的增加而线性增加, 该线性关系适用于所有双层R-P结构A3B2O7的HIF[118].本课题组[84]通过高能球磨提高了煅烧粉末的烧结活性, 在高质量的致密Sr3?xBaxSn2O7陶瓷中获得了0.26 μC/cm2的铁电极化强度和260 kV/cm的矫顽场, 与Sr3Sn2O7陶瓷相比, 铁电性能有所改善, 这归因于τ的降低.Sr3Sn2O7陶瓷的铁电极化强度为0.08 μC/cm2, 矫顽场为335 kV/cm.由于Ca3Sn2O7具有较大的理论极化强度, 在Sr3Sn2O7陶瓷的A位引入Ca2+可以有效改善铁电性, 且TC随着Ca2+含量增加而升高[85].在这些样品中, Sr3Sn2O7单晶表现出最出色的铁电性能, 沿[100]面测得的铁电极化强度为1.1 μC/cm2, 矫顽场为39 kV/cm[112].典型双层R-P结构Sr基氧化物的铁电性能对比如图8所示, 基于Sr3Sn2O7陶瓷进行A位置换得到的(Sr,Ca)3Sn2O7陶瓷的矫顽场从335 kV/cm降低至200 kV/cm左右, 铁电极化强度与Sr3Sn2O7陶瓷相当[85], 而A位置换的(Sr,Ba)3Sn2O7陶瓷相对Sr3Sn2O7陶瓷而言获得了较高的铁电极化强度[84].基于Sr3Zr2O7陶瓷的A位置换提高了(Sr,Ba)3Zr2O7陶瓷的铁电极化强度, 但矫顽场显著增高, 而B位置换的Sr3(Sn,Zr)2O7陶瓷的铁电性能与Sr3Zr2O7陶瓷较为接近. ... The first room-temperature ferroelectric Sn insulator and its polarization switching kinetics 2 2017 ... Sr3Sn2O7氧化物具有双层R-P结构, 其晶格参数为a=5.710 ?、b=5.736 ?和c=20.688 ?[114].尽管早期中子粉末衍射(NPD)实验显示, Sr3Sn2O7属于正交的Cmcm空间群[115], 但可能通过复杂晶格畸变的冻结而诱发HIF[15,26,49,116].2017年, Wang等[117]在双层R-P结构的Sr3Sn2O7陶瓷中首次观测到HIF, 利用XRD精修确定了Sr3Sn2O7在室温下属于非中心对称极性空间群A21am, 并通过PUND法获得了典型的室温电滞回线, 其剩余极化强度和矫顽场分别为0.1 μC/cm2和200 kV/cm.随后, Xu等[112]报道称Sr3Sn2O7单晶沿[100]的矫顽场最低可达39 kV/cm, 这是目前已知HIF体系中最小的室温矫顽场, 与IP-PFM观测到带电DWs和中性DWs有关.结合Yoshida等[118]和本课题组的理论计算与实验数据, Sr3Sn2O7的相变历程为I4/mmm→Acaa→Pnab→A21am.Yoshida和本课题组总结了双层R-P材料的TC与τ呈现线性负相关的关系, 发现TC随着Ca2+含量的增加而线性增加, 该线性关系适用于所有双层R-P结构A3B2O7的HIF[118].本课题组[84]通过高能球磨提高了煅烧粉末的烧结活性, 在高质量的致密Sr3?xBaxSn2O7陶瓷中获得了0.26 μC/cm2的铁电极化强度和260 kV/cm的矫顽场, 与Sr3Sn2O7陶瓷相比, 铁电性能有所改善, 这归因于τ的降低.Sr3Sn2O7陶瓷的铁电极化强度为0.08 μC/cm2, 矫顽场为335 kV/cm.由于Ca3Sn2O7具有较大的理论极化强度, 在Sr3Sn2O7陶瓷的A位引入Ca2+可以有效改善铁电性, 且TC随着Ca2+含量增加而升高[85].在这些样品中, Sr3Sn2O7单晶表现出最出色的铁电性能, 沿[100]面测得的铁电极化强度为1.1 μC/cm2, 矫顽场为39 kV/cm[112].典型双层R-P结构Sr基氧化物的铁电性能对比如图8所示, 基于Sr3Sn2O7陶瓷进行A位置换得到的(Sr,Ca)3Sn2O7陶瓷的矫顽场从335 kV/cm降低至200 kV/cm左右, 铁电极化强度与Sr3Sn2O7陶瓷相当[85], 而A位置换的(Sr,Ba)3Sn2O7陶瓷相对Sr3Sn2O7陶瓷而言获得了较高的铁电极化强度[84].基于Sr3Zr2O7陶瓷的A位置换提高了(Sr,Ba)3Zr2O7陶瓷的铁电极化强度, 但矫顽场显著增高, 而B位置换的Sr3(Sn,Zr)2O7陶瓷的铁电性能与Sr3Zr2O7陶瓷较为接近. ...
由DFT计算可知, τ对 TC和铁电性能有显著影响[ 26]. 图10总结了典型的双层R-P结构HIF材料中 τ(此处使用Shannon离子半径[ 118])和 TC之间的关系, 包括(Sr,Ca) 3Ti 2O 7陶瓷[ 73]、(Ca,La) 3(Ti,Al) 2O 7陶瓷[ 64]、Ca 3(Mn,Ti) 2O 7陶瓷[ 43]、Ca 3[Mn 1?x(Fe 0.5Nb 0.5) x] 2O 7陶瓷[ 45]、Ca 3(Mn,Ti) 2O 7陶瓷[ 14]、(Sr,Ba) 3Sn 2O 7陶瓷[ 84]、(Sr,Ca) 3Sn 2O 7陶瓷[ 85]、(Sr,Ba) 3Zr 2O 7陶瓷[ 86]和La 2Sr(Sc,Fe) 2O 7陶瓷[ 121].红色拟合线与文献[ 118]报道的结果一致, 双层R-P结构HIF材料中 τ与 TC呈负相关.已报道的双层R-P结构氧化物的 τ均小于1, 而类钙钛矿结构材料的 τ偏离标准钙钛矿(1)越大, 氧八面体畸变幅度越大, 即铁电相变所需克服的能垒越高, 从而导致铁电相变的 TC升高.然而, 在(Ca,La) 3(Ti,Al) 2O 7陶瓷中观察到发散行为, 表明仅考虑 τ的平均尺寸效应不够充分[ 122].对于Ca 3Ti 2O 7化合物, 该结构由带电荷中性的TiO 2、CaO AO层和ABO 3层沿[001]方向堆叠形成, 而(Ca,La)O钙钛矿层和(Ti,Al)O 2中将异价La3+和Al3+阳离子分别引入Ca 3Ti 2O 7的A位和B位点后分别带正电荷和负电荷[ 122].在此情况下, 正离子置换可以导致氧八面体畸变幅度减小, 符合Sr基陶瓷的研究结果[ 86].异价取代导致氧八面体畸变幅度急剧下降, 进而使铁电 TC急剧下降.基于上述分析, 异价取代可能削弱HIF材料的铁电性能.这一点在后面的La 2SrSc 2O 7基陶瓷中得到验证, 这是因为异价置换升高层间电荷, 引起层间褶皱, 抑制了 a0 a0 c+旋转, 降低了材料的 TC[ 121, 123]. ... Hybrid improper ferroelectricity and possible ferroelectric switching paths in Sr3Hf2O7 3 2019 ... 为了拓展HIF的研究, Yoshida等[119]发现Sr3Zr2O7也具有HIF, 其铁电性起源于氧八面体倾侧和旋转以及自发极化之间的三线耦合机制, 经历的相变过程为I4/mmm→Amam→Pnab→A21am.结合SHG信号、同步辐射XRD和NPD结果, 确定Sr3Zr2O7属于极性A21am空间群, 并根据NPD精修结果计算发现晶体结构分层极化, 如图9(c)所示.Sr3Zr2O7陶瓷在室温下获得的铁电极化强度和矫顽场分别为0.3 μC/cm2和150 kV/cm[119].SHG信号表明Sr3Zr2O7中存在非中心对称结构, 并结合同步辐射XRD和NPD分析结果证明了极性A21am结构.Sr3Zr2O7陶瓷的剩余极化强度和矫顽场分别达到0.3 μC/cm2和150 kV/cm.其铁电极化强度与Ca3Ti2O7陶瓷 (0.6 μC/cm2)[43]和Sr3Sn2O7陶瓷(0.25 μC/cm2)[117]相当, 但比DFT计算的铁电极化强度(6.75 μC/cm2)低一个数量级.虽然在(Sr,Ba)3Zr2O7陶瓷中获得了更高的铁电极化强度, 但其矫顽场仍未得到有效控制[86].Sr3M2O7化合物普遍经历复杂的相变过程, I4/mmm到A21am的相变过程中经历了两次二级相变(图9(a, b))[118], 这种独特的铁电相变路径可能源于材料中多种模式的非谐相互作用, 通过计算铁电翻转路径的最低能量, 可以对复杂的相变历程有更深入的理解.这个问题可以通过识别具有最小能量路径的铁电翻转路径来解决[59-60].基于此, 本课题组[120]通过DFT计算揭示了Sr3Hf2O7的HIF和铁电翻转路径, 发现从Sr3Hf2O7非极性Pbnm到Pnab相的四种相变路径的能量势垒几乎相同, 这为Sr基氧化物的复杂相变提供新思路, 而Sr3Hf2O7相对较低的理论极化强度是由HfO2层的负贡献引起, 如图9(d)所示. ...
尽管理论上已经预测并成功合成了许多具有单层R-P结构的非中心对称结构氧化物, 并对部分单层R-P氧化物的压电特性进行了表征, 但其可翻转的铁电极化鲜有报道[ 139- 140, 148].本课题组通过放电等离子烧结工艺成功制备了致密的Li 2La 2Ti 3O 10陶瓷, 并利用PUND法在室温下观测到了其饱和电滞回线, 测得剩余极化强度为0.25 μC/cm2, 矫顽场为260 kV/cm[ 149], 如 图16(a)所示.结合NPD, 同步辐射XRD和选区电子衍射(SAED)确定Li 2La 2Ti 3O 10陶瓷具有极性P2 1ab空间群的三层R-P结构[ 149].此外, DFT计算结果显示Pc、P2 1ab、P2 1am(No. 26)、A2 1am和P2 1an空间群与Li 2La 2Ti 3O 10结构精修的拟合优度χ2分别为6.15、4.82、5.12、4.95、5.43, 加权图形剩余方差因子 Rwp分别为9.19%、8.16%、8.41%、8.30%、8.66%, 进一步验证了Li 2La 2Ti 3O 10氧化物形成能最低的空间群为P2 1ab( 图16(b)).这项工作首次在实验上证实了A位阳离子有序的三层R-P结构的Li 2La 2Ti 3O 10陶瓷在室温下具有铁电性, 极大地扩展了HIF的研究领域[ 149].随后, 本课题组基于NPD数据精修确定Li 2Nd 2Ti 3O 10陶瓷在室温下属于极性P2 1ab相[ 150].PUND法测得其饱和室温电滞回线, 具有约0.4 μC/cm2的剩余极化强度和150 kV/cm的矫顽场, 如 图16(c)所示.Li 2Nd 2Ti 3O 10陶瓷的铁电性同样起源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的协同作用[ 150].与Li 2La 2Ti 3O 10陶瓷相比, Li 2Nd 2Ti 3O 10陶瓷表现出更高的铁电极化强度和较低的矫顽场, 这可能是源于Nd3+离子半径小于La3+导致更显著的氧八面体畸变( 图16(d)), 进而获得了更优异的铁电性能.而较低的矫顽场则与Li 2La 2Ti 3O 10陶瓷含有少量的第二相有关.Li 2La 2Ti 3O 10陶瓷中的铁电性源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的耦合, 其中A位阳离子有序是奇数层R-P结构材料获得极性相的关键因素[ 149], 其晶体结构如 图16(e)所示. 图16(f)展示了R-P层状结构中各层极化对宏观极化的贡献, 宏观极化主要源于TiO 6氧八面体畸变, 这与双层R-P氧化物的铁电起源完全不同; 双层R-P氧化物的宏观极化是由A位阳离子的未抵消反铁畸变位移引起.A位离子有序三层R-P结构陶瓷的实验工作为进一步拓宽HIF的研究宽度与广度提供了新的见解, 且研究思路可以拓展到其余层状钙钛矿氧化物[ 149- 150]. ...
尽管理论上已经预测并成功合成了许多具有单层R-P结构的非中心对称结构氧化物, 并对部分单层R-P氧化物的压电特性进行了表征, 但其可翻转的铁电极化鲜有报道[ 139- 140, 148].本课题组通过放电等离子烧结工艺成功制备了致密的Li 2La 2Ti 3O 10陶瓷, 并利用PUND法在室温下观测到了其饱和电滞回线, 测得剩余极化强度为0.25 μC/cm2, 矫顽场为260 kV/cm[ 149], 如 图16(a)所示.结合NPD, 同步辐射XRD和选区电子衍射(SAED)确定Li 2La 2Ti 3O 10陶瓷具有极性P2 1ab空间群的三层R-P结构[ 149].此外, DFT计算结果显示Pc、P2 1ab、P2 1am(No. 26)、A2 1am和P2 1an空间群与Li 2La 2Ti 3O 10结构精修的拟合优度χ2分别为6.15、4.82、5.12、4.95、5.43, 加权图形剩余方差因子 Rwp分别为9.19%、8.16%、8.41%、8.30%、8.66%, 进一步验证了Li 2La 2Ti 3O 10氧化物形成能最低的空间群为P2 1ab( 图16(b)).这项工作首次在实验上证实了A位阳离子有序的三层R-P结构的Li 2La 2Ti 3O 10陶瓷在室温下具有铁电性, 极大地扩展了HIF的研究领域[ 149].随后, 本课题组基于NPD数据精修确定Li 2Nd 2Ti 3O 10陶瓷在室温下属于极性P2 1ab相[ 150].PUND法测得其饱和室温电滞回线, 具有约0.4 μC/cm2的剩余极化强度和150 kV/cm的矫顽场, 如 图16(c)所示.Li 2Nd 2Ti 3O 10陶瓷的铁电性同样起源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的协同作用[ 150].与Li 2La 2Ti 3O 10陶瓷相比, Li 2Nd 2Ti 3O 10陶瓷表现出更高的铁电极化强度和较低的矫顽场, 这可能是源于Nd3+离子半径小于La3+导致更显著的氧八面体畸变( 图16(d)), 进而获得了更优异的铁电性能.而较低的矫顽场则与Li 2La 2Ti 3O 10陶瓷含有少量的第二相有关.Li 2La 2Ti 3O 10陶瓷中的铁电性源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的耦合, 其中A位阳离子有序是奇数层R-P结构材料获得极性相的关键因素[ 149], 其晶体结构如 图16(e)所示. 图16(f)展示了R-P层状结构中各层极化对宏观极化的贡献, 宏观极化主要源于TiO 6氧八面体畸变, 这与双层R-P氧化物的铁电起源完全不同; 双层R-P氧化物的宏观极化是由A位阳离子的未抵消反铁畸变位移引起.A位离子有序三层R-P结构陶瓷的实验工作为进一步拓宽HIF的研究宽度与广度提供了新的见解, 且研究思路可以拓展到其余层状钙钛矿氧化物[ 149- 150]. ...
尽管理论上已经预测并成功合成了许多具有单层R-P结构的非中心对称结构氧化物, 并对部分单层R-P氧化物的压电特性进行了表征, 但其可翻转的铁电极化鲜有报道[ 139- 140, 148].本课题组通过放电等离子烧结工艺成功制备了致密的Li 2La 2Ti 3O 10陶瓷, 并利用PUND法在室温下观测到了其饱和电滞回线, 测得剩余极化强度为0.25 μC/cm2, 矫顽场为260 kV/cm[ 149], 如 图16(a)所示.结合NPD, 同步辐射XRD和选区电子衍射(SAED)确定Li 2La 2Ti 3O 10陶瓷具有极性P2 1ab空间群的三层R-P结构[ 149].此外, DFT计算结果显示Pc、P2 1ab、P2 1am(No. 26)、A2 1am和P2 1an空间群与Li 2La 2Ti 3O 10结构精修的拟合优度χ2分别为6.15、4.82、5.12、4.95、5.43, 加权图形剩余方差因子 Rwp分别为9.19%、8.16%、8.41%、8.30%、8.66%, 进一步验证了Li 2La 2Ti 3O 10氧化物形成能最低的空间群为P2 1ab( 图16(b)).这项工作首次在实验上证实了A位阳离子有序的三层R-P结构的Li 2La 2Ti 3O 10陶瓷在室温下具有铁电性, 极大地扩展了HIF的研究领域[ 149].随后, 本课题组基于NPD数据精修确定Li 2Nd 2Ti 3O 10陶瓷在室温下属于极性P2 1ab相[ 150].PUND法测得其饱和室温电滞回线, 具有约0.4 μC/cm2的剩余极化强度和150 kV/cm的矫顽场, 如 图16(c)所示.Li 2Nd 2Ti 3O 10陶瓷的铁电性同样起源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的协同作用[ 150].与Li 2La 2Ti 3O 10陶瓷相比, Li 2Nd 2Ti 3O 10陶瓷表现出更高的铁电极化强度和较低的矫顽场, 这可能是源于Nd3+离子半径小于La3+导致更显著的氧八面体畸变( 图16(d)), 进而获得了更优异的铁电性能.而较低的矫顽场则与Li 2La 2Ti 3O 10陶瓷含有少量的第二相有关.Li 2La 2Ti 3O 10陶瓷中的铁电性源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的耦合, 其中A位阳离子有序是奇数层R-P结构材料获得极性相的关键因素[ 149], 其晶体结构如 图16(e)所示. 图16(f)展示了R-P层状结构中各层极化对宏观极化的贡献, 宏观极化主要源于TiO 6氧八面体畸变, 这与双层R-P氧化物的铁电起源完全不同; 双层R-P氧化物的宏观极化是由A位阳离子的未抵消反铁畸变位移引起.A位离子有序三层R-P结构陶瓷的实验工作为进一步拓宽HIF的研究宽度与广度提供了新的见解, 且研究思路可以拓展到其余层状钙钛矿氧化物[ 149- 150]. ...
尽管理论上已经预测并成功合成了许多具有单层R-P结构的非中心对称结构氧化物, 并对部分单层R-P氧化物的压电特性进行了表征, 但其可翻转的铁电极化鲜有报道[ 139- 140, 148].本课题组通过放电等离子烧结工艺成功制备了致密的Li 2La 2Ti 3O 10陶瓷, 并利用PUND法在室温下观测到了其饱和电滞回线, 测得剩余极化强度为0.25 μC/cm2, 矫顽场为260 kV/cm[ 149], 如 图16(a)所示.结合NPD, 同步辐射XRD和选区电子衍射(SAED)确定Li 2La 2Ti 3O 10陶瓷具有极性P2 1ab空间群的三层R-P结构[ 149].此外, DFT计算结果显示Pc、P2 1ab、P2 1am(No. 26)、A2 1am和P2 1an空间群与Li 2La 2Ti 3O 10结构精修的拟合优度χ2分别为6.15、4.82、5.12、4.95、5.43, 加权图形剩余方差因子 Rwp分别为9.19%、8.16%、8.41%、8.30%、8.66%, 进一步验证了Li 2La 2Ti 3O 10氧化物形成能最低的空间群为P2 1ab( 图16(b)).这项工作首次在实验上证实了A位阳离子有序的三层R-P结构的Li 2La 2Ti 3O 10陶瓷在室温下具有铁电性, 极大地扩展了HIF的研究领域[ 149].随后, 本课题组基于NPD数据精修确定Li 2Nd 2Ti 3O 10陶瓷在室温下属于极性P2 1ab相[ 150].PUND法测得其饱和室温电滞回线, 具有约0.4 μC/cm2的剩余极化强度和150 kV/cm的矫顽场, 如 图16(c)所示.Li 2Nd 2Ti 3O 10陶瓷的铁电性同样起源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的协同作用[ 150].与Li 2La 2Ti 3O 10陶瓷相比, Li 2Nd 2Ti 3O 10陶瓷表现出更高的铁电极化强度和较低的矫顽场, 这可能是源于Nd3+离子半径小于La3+导致更显著的氧八面体畸变( 图16(d)), 进而获得了更优异的铁电性能.而较低的矫顽场则与Li 2La 2Ti 3O 10陶瓷含有少量的第二相有关.Li 2La 2Ti 3O 10陶瓷中的铁电性源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的耦合, 其中A位阳离子有序是奇数层R-P结构材料获得极性相的关键因素[ 149], 其晶体结构如 图16(e)所示. 图16(f)展示了R-P层状结构中各层极化对宏观极化的贡献, 宏观极化主要源于TiO 6氧八面体畸变, 这与双层R-P氧化物的铁电起源完全不同; 双层R-P氧化物的宏观极化是由A位阳离子的未抵消反铁畸变位移引起.A位离子有序三层R-P结构陶瓷的实验工作为进一步拓宽HIF的研究宽度与广度提供了新的见解, 且研究思路可以拓展到其余层状钙钛矿氧化物[ 149- 150]. ... Inversion symmetry breaking by oxygen octahedral rotations in the Ruddlesden-Popper NaRTiO4 family 4 2014 ... 前人的理论计算表明, R-P型层状钙钛矿材料中, 若A位离子呈有序排列, 则其铁电性有可能增强[26].更为重要的是, 在单层或三层等奇数层R-P材料中也可能出现铁电性, 而在A位离子无序状态下, 这是难以实现的(当n为奇数时, 材料具有n+1偶数层的A位离子, 在完全无序的情况下, 其反铁畸变位移将被完全抵消), 比如具有Pbca空间群的Ca4Ti3O10就属于非极性相[139-142].因此, 研究A位离子有序的R-P材料能有效地拓宽HIF的研究范围.同时, 由于对称性在奇数和偶数层R-P材料中有所差异, 其铁电性起源也可能不同, 因此研究奇数层R-P材料有助于加深对HIF物理本质的理解.2004年, Rodgers等[143]首先利用中子和电子衍射发现了A、B位离子同时有序的单层R-P材料La2Sr2LiRuO8, 其晶体结构如图15(a)所示, 并确认其室温结构为极性Imm2正交相, 但其宏观铁电极化尚未报道.Akamatsu课题组[139-142]借助同步辐射XRD和DFT计算, 研究了A位离子有序单层R-P材料MRTiO4(M=H、Li、Na、K, R为稀土)的晶体结构, 发现氧八面体的倾转破坏了材料的反演对称性, 仅形成了非中心对称的非极性P$\bar{4}$21m四方相.LiRTiO4和NaRTiO4化合物的非中心对称起源于氧八面体的a?b0c0/b0a?c0旋转, 其中LiRTiO4中形成了LiO反萤石层, 而NaRTiO4中形成的是NaO岩盐层(图15(b)), 并且氧八面体畸变幅度的大小取决于稀土阳离子的离子半径[139,142].HRTiO4是单层R-P结构系列的衍生物, 其中TiO6八面体层之间的HO层在结构和化学环境上与HRTiO4[139]和NaRTiO4[142]中的LiO和NaO层完全不同.早期文献指出HRTiO4(R=La、Nd)属于P4/nmm空间群[144], HYTiO4属于P21/c空间群[145], HSmTiO4属于I4/mmm空间群[146].此外, Silyukov等[147]发现HRTiO4(R=La、Nd)氧化物在298~1273 K之间经历了结构相变和脱水.HRTiO4氧化物具有中心对称结构, 可以作为压电材料应用于光催化领域.实际上, 优化稀土离子配位环境是氧八面体旋转的主要驱动力[147].相反, AARTiO4(AA=Na、K、Rb)碱金属离子对氧八面体旋转的影响较小, 其作用是施加相邻层的“键应变”.因此, 层间晶格失配解释了A位碱金属尺寸对八面体旋转不稳定性的影响.其SHG信号随温度变化的曲线可分辨出AARTiO4(AA=Na、K、Rb)氧化物属于非中心对称结构, 如图15(c)所示[141].值得注意的是, 稀土离子的配位环境在这些化合物中诱导了氧八面体畸变: 离子半径较小的稀土阳离子与氧离子键合较弱, 驱动氧八面体较大畸变; 而离子半径较大的稀土阳离子键合过强, 氧八面体畸变发生的驱动力较弱.通过调节A位碱金属离子尺寸可实现氧八面体畸变幅度和宏观铁电性的可控调节[141].此外,当没有氧八面体畸变时, 质子层会发生层间滑移, 从而优化了氢键在晶体结构中的键长和键方向[140]. ...
尽管理论上已经预测并成功合成了许多具有单层R-P结构的非中心对称结构氧化物, 并对部分单层R-P氧化物的压电特性进行了表征, 但其可翻转的铁电极化鲜有报道[ 139- 140, 148].本课题组通过放电等离子烧结工艺成功制备了致密的Li 2La 2Ti 3O 10陶瓷, 并利用PUND法在室温下观测到了其饱和电滞回线, 测得剩余极化强度为0.25 μC/cm2, 矫顽场为260 kV/cm[ 149], 如 图16(a)所示.结合NPD, 同步辐射XRD和选区电子衍射(SAED)确定Li 2La 2Ti 3O 10陶瓷具有极性P2 1ab空间群的三层R-P结构[ 149].此外, DFT计算结果显示Pc、P2 1ab、P2 1am(No. 26)、A2 1am和P2 1an空间群与Li 2La 2Ti 3O 10结构精修的拟合优度χ2分别为6.15、4.82、5.12、4.95、5.43, 加权图形剩余方差因子 Rwp分别为9.19%、8.16%、8.41%、8.30%、8.66%, 进一步验证了Li 2La 2Ti 3O 10氧化物形成能最低的空间群为P2 1ab( 图16(b)).这项工作首次在实验上证实了A位阳离子有序的三层R-P结构的Li 2La 2Ti 3O 10陶瓷在室温下具有铁电性, 极大地扩展了HIF的研究领域[ 149].随后, 本课题组基于NPD数据精修确定Li 2Nd 2Ti 3O 10陶瓷在室温下属于极性P2 1ab相[ 150].PUND法测得其饱和室温电滞回线, 具有约0.4 μC/cm2的剩余极化强度和150 kV/cm的矫顽场, 如 图16(c)所示.Li 2Nd 2Ti 3O 10陶瓷的铁电性同样起源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的协同作用[ 150].与Li 2La 2Ti 3O 10陶瓷相比, Li 2Nd 2Ti 3O 10陶瓷表现出更高的铁电极化强度和较低的矫顽场, 这可能是源于Nd3+离子半径小于La3+导致更显著的氧八面体畸变( 图16(d)), 进而获得了更优异的铁电性能.而较低的矫顽场则与Li 2La 2Ti 3O 10陶瓷含有少量的第二相有关.Li 2La 2Ti 3O 10陶瓷中的铁电性源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的耦合, 其中A位阳离子有序是奇数层R-P结构材料获得极性相的关键因素[ 149], 其晶体结构如 图16(e)所示. 图16(f)展示了R-P层状结构中各层极化对宏观极化的贡献, 宏观极化主要源于TiO 6氧八面体畸变, 这与双层R-P氧化物的铁电起源完全不同; 双层R-P氧化物的宏观极化是由A位阳离子的未抵消反铁畸变位移引起.A位离子有序三层R-P结构陶瓷的实验工作为进一步拓宽HIF的研究宽度与广度提供了新的见解, 且研究思路可以拓展到其余层状钙钛矿氧化物[ 149- 150]. ...
尽管理论上已经预测并成功合成了许多具有单层R-P结构的非中心对称结构氧化物, 并对部分单层R-P氧化物的压电特性进行了表征, 但其可翻转的铁电极化鲜有报道[ 139- 140, 148].本课题组通过放电等离子烧结工艺成功制备了致密的Li 2La 2Ti 3O 10陶瓷, 并利用PUND法在室温下观测到了其饱和电滞回线, 测得剩余极化强度为0.25 μC/cm2, 矫顽场为260 kV/cm[ 149], 如 图16(a)所示.结合NPD, 同步辐射XRD和选区电子衍射(SAED)确定Li 2La 2Ti 3O 10陶瓷具有极性P2 1ab空间群的三层R-P结构[ 149].此外, DFT计算结果显示Pc、P2 1ab、P2 1am(No. 26)、A2 1am和P2 1an空间群与Li 2La 2Ti 3O 10结构精修的拟合优度χ2分别为6.15、4.82、5.12、4.95、5.43, 加权图形剩余方差因子 Rwp分别为9.19%、8.16%、8.41%、8.30%、8.66%, 进一步验证了Li 2La 2Ti 3O 10氧化物形成能最低的空间群为P2 1ab( 图16(b)).这项工作首次在实验上证实了A位阳离子有序的三层R-P结构的Li 2La 2Ti 3O 10陶瓷在室温下具有铁电性, 极大地扩展了HIF的研究领域[ 149].随后, 本课题组基于NPD数据精修确定Li 2Nd 2Ti 3O 10陶瓷在室温下属于极性P2 1ab相[ 150].PUND法测得其饱和室温电滞回线, 具有约0.4 μC/cm2的剩余极化强度和150 kV/cm的矫顽场, 如 图16(c)所示.Li 2Nd 2Ti 3O 10陶瓷的铁电性同样起源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的协同作用[ 150].与Li 2La 2Ti 3O 10陶瓷相比, Li 2Nd 2Ti 3O 10陶瓷表现出更高的铁电极化强度和较低的矫顽场, 这可能是源于Nd3+离子半径小于La3+导致更显著的氧八面体畸变( 图16(d)), 进而获得了更优异的铁电性能.而较低的矫顽场则与Li 2La 2Ti 3O 10陶瓷含有少量的第二相有关.Li 2La 2Ti 3O 10陶瓷中的铁电性源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的耦合, 其中A位阳离子有序是奇数层R-P结构材料获得极性相的关键因素[ 149], 其晶体结构如 图16(e)所示. 图16(f)展示了R-P层状结构中各层极化对宏观极化的贡献, 宏观极化主要源于TiO 6氧八面体畸变, 这与双层R-P氧化物的铁电起源完全不同; 双层R-P氧化物的宏观极化是由A位阳离子的未抵消反铁畸变位移引起.A位离子有序三层R-P结构陶瓷的实验工作为进一步拓宽HIF的研究宽度与广度提供了新的见解, 且研究思路可以拓展到其余层状钙钛矿氧化物[ 149- 150]. ... Structure determination of n=1 Ruddlesden-Popper compound HLaTiO4 by powder neutron diffraction 1 2006 ... 前人的理论计算表明, R-P型层状钙钛矿材料中, 若A位离子呈有序排列, 则其铁电性有可能增强[26].更为重要的是, 在单层或三层等奇数层R-P材料中也可能出现铁电性, 而在A位离子无序状态下, 这是难以实现的(当n为奇数时, 材料具有n+1偶数层的A位离子, 在完全无序的情况下, 其反铁畸变位移将被完全抵消), 比如具有Pbca空间群的Ca4Ti3O10就属于非极性相[139-142].因此, 研究A位离子有序的R-P材料能有效地拓宽HIF的研究范围.同时, 由于对称性在奇数和偶数层R-P材料中有所差异, 其铁电性起源也可能不同, 因此研究奇数层R-P材料有助于加深对HIF物理本质的理解.2004年, Rodgers等[143]首先利用中子和电子衍射发现了A、B位离子同时有序的单层R-P材料La2Sr2LiRuO8, 其晶体结构如图15(a)所示, 并确认其室温结构为极性Imm2正交相, 但其宏观铁电极化尚未报道.Akamatsu课题组[139-142]借助同步辐射XRD和DFT计算, 研究了A位离子有序单层R-P材料MRTiO4(M=H、Li、Na、K, R为稀土)的晶体结构, 发现氧八面体的倾转破坏了材料的反演对称性, 仅形成了非中心对称的非极性P$\bar{4}$21m四方相.LiRTiO4和NaRTiO4化合物的非中心对称起源于氧八面体的a?b0c0/b0a?c0旋转, 其中LiRTiO4中形成了LiO反萤石层, 而NaRTiO4中形成的是NaO岩盐层(图15(b)), 并且氧八面体畸变幅度的大小取决于稀土阳离子的离子半径[139,142].HRTiO4是单层R-P结构系列的衍生物, 其中TiO6八面体层之间的HO层在结构和化学环境上与HRTiO4[139]和NaRTiO4[142]中的LiO和NaO层完全不同.早期文献指出HRTiO4(R=La、Nd)属于P4/nmm空间群[144], HYTiO4属于P21/c空间群[145], HSmTiO4属于I4/mmm空间群[146].此外, Silyukov等[147]发现HRTiO4(R=La、Nd)氧化物在298~1273 K之间经历了结构相变和脱水.HRTiO4氧化物具有中心对称结构, 可以作为压电材料应用于光催化领域.实际上, 优化稀土离子配位环境是氧八面体旋转的主要驱动力[147].相反, AARTiO4(AA=Na、K、Rb)碱金属离子对氧八面体旋转的影响较小, 其作用是施加相邻层的“键应变”.因此, 层间晶格失配解释了A位碱金属尺寸对八面体旋转不稳定性的影响.其SHG信号随温度变化的曲线可分辨出AARTiO4(AA=Na、K、Rb)氧化物属于非中心对称结构, 如图15(c)所示[141].值得注意的是, 稀土离子的配位环境在这些化合物中诱导了氧八面体畸变: 离子半径较小的稀土阳离子与氧离子键合较弱, 驱动氧八面体较大畸变; 而离子半径较大的稀土阳离子键合过强, 氧八面体畸变发生的驱动力较弱.通过调节A位碱金属离子尺寸可实现氧八面体畸变幅度和宏观铁电性的可控调节[141].此外,当没有氧八面体畸变时, 质子层会发生层间滑移, 从而优化了氢键在晶体结构中的键长和键方向[140]. ... Structural change in a series of protonated layered perovskite compounds, HLnTiO4 (Ln=La, Nd and Y) 1 2006 ... 前人的理论计算表明, R-P型层状钙钛矿材料中, 若A位离子呈有序排列, 则其铁电性有可能增强[26].更为重要的是, 在单层或三层等奇数层R-P材料中也可能出现铁电性, 而在A位离子无序状态下, 这是难以实现的(当n为奇数时, 材料具有n+1偶数层的A位离子, 在完全无序的情况下, 其反铁畸变位移将被完全抵消), 比如具有Pbca空间群的Ca4Ti3O10就属于非极性相[139-142].因此, 研究A位离子有序的R-P材料能有效地拓宽HIF的研究范围.同时, 由于对称性在奇数和偶数层R-P材料中有所差异, 其铁电性起源也可能不同, 因此研究奇数层R-P材料有助于加深对HIF物理本质的理解.2004年, Rodgers等[143]首先利用中子和电子衍射发现了A、B位离子同时有序的单层R-P材料La2Sr2LiRuO8, 其晶体结构如图15(a)所示, 并确认其室温结构为极性Imm2正交相, 但其宏观铁电极化尚未报道.Akamatsu课题组[139-142]借助同步辐射XRD和DFT计算, 研究了A位离子有序单层R-P材料MRTiO4(M=H、Li、Na、K, R为稀土)的晶体结构, 发现氧八面体的倾转破坏了材料的反演对称性, 仅形成了非中心对称的非极性P$\bar{4}$21m四方相.LiRTiO4和NaRTiO4化合物的非中心对称起源于氧八面体的a?b0c0/b0a?c0旋转, 其中LiRTiO4中形成了LiO反萤石层, 而NaRTiO4中形成的是NaO岩盐层(图15(b)), 并且氧八面体畸变幅度的大小取决于稀土阳离子的离子半径[139,142].HRTiO4是单层R-P结构系列的衍生物, 其中TiO6八面体层之间的HO层在结构和化学环境上与HRTiO4[139]和NaRTiO4[142]中的LiO和NaO层完全不同.早期文献指出HRTiO4(R=La、Nd)属于P4/nmm空间群[144], HYTiO4属于P21/c空间群[145], HSmTiO4属于I4/mmm空间群[146].此外, Silyukov等[147]发现HRTiO4(R=La、Nd)氧化物在298~1273 K之间经历了结构相变和脱水.HRTiO4氧化物具有中心对称结构, 可以作为压电材料应用于光催化领域.实际上, 优化稀土离子配位环境是氧八面体旋转的主要驱动力[147].相反, AARTiO4(AA=Na、K、Rb)碱金属离子对氧八面体旋转的影响较小, 其作用是施加相邻层的“键应变”.因此, 层间晶格失配解释了A位碱金属尺寸对八面体旋转不稳定性的影响.其SHG信号随温度变化的曲线可分辨出AARTiO4(AA=Na、K、Rb)氧化物属于非中心对称结构, 如图15(c)所示[141].值得注意的是, 稀土离子的配位环境在这些化合物中诱导了氧八面体畸变: 离子半径较小的稀土阳离子与氧离子键合较弱, 驱动氧八面体较大畸变; 而离子半径较大的稀土阳离子键合过强, 氧八面体畸变发生的驱动力较弱.通过调节A位碱金属离子尺寸可实现氧八面体畸变幅度和宏观铁电性的可控调节[141].此外,当没有氧八面体畸变时, 质子层会发生层间滑移, 从而优化了氢键在晶体结构中的键长和键方向[140]. ... A new family of protonated oxides HLnTiO4 (Ln=La, Nd, Sm, and Gd) 1 1996 ... 前人的理论计算表明, R-P型层状钙钛矿材料中, 若A位离子呈有序排列, 则其铁电性有可能增强[26].更为重要的是, 在单层或三层等奇数层R-P材料中也可能出现铁电性, 而在A位离子无序状态下, 这是难以实现的(当n为奇数时, 材料具有n+1偶数层的A位离子, 在完全无序的情况下, 其反铁畸变位移将被完全抵消), 比如具有Pbca空间群的Ca4Ti3O10就属于非极性相[139-142].因此, 研究A位离子有序的R-P材料能有效地拓宽HIF的研究范围.同时, 由于对称性在奇数和偶数层R-P材料中有所差异, 其铁电性起源也可能不同, 因此研究奇数层R-P材料有助于加深对HIF物理本质的理解.2004年, Rodgers等[143]首先利用中子和电子衍射发现了A、B位离子同时有序的单层R-P材料La2Sr2LiRuO8, 其晶体结构如图15(a)所示, 并确认其室温结构为极性Imm2正交相, 但其宏观铁电极化尚未报道.Akamatsu课题组[139-142]借助同步辐射XRD和DFT计算, 研究了A位离子有序单层R-P材料MRTiO4(M=H、Li、Na、K, R为稀土)的晶体结构, 发现氧八面体的倾转破坏了材料的反演对称性, 仅形成了非中心对称的非极性P$\bar{4}$21m四方相.LiRTiO4和NaRTiO4化合物的非中心对称起源于氧八面体的a?b0c0/b0a?c0旋转, 其中LiRTiO4中形成了LiO反萤石层, 而NaRTiO4中形成的是NaO岩盐层(图15(b)), 并且氧八面体畸变幅度的大小取决于稀土阳离子的离子半径[139,142].HRTiO4是单层R-P结构系列的衍生物, 其中TiO6八面体层之间的HO层在结构和化学环境上与HRTiO4[139]和NaRTiO4[142]中的LiO和NaO层完全不同.早期文献指出HRTiO4(R=La、Nd)属于P4/nmm空间群[144], HYTiO4属于P21/c空间群[145], HSmTiO4属于I4/mmm空间群[146].此外, Silyukov等[147]发现HRTiO4(R=La、Nd)氧化物在298~1273 K之间经历了结构相变和脱水.HRTiO4氧化物具有中心对称结构, 可以作为压电材料应用于光催化领域.实际上, 优化稀土离子配位环境是氧八面体旋转的主要驱动力[147].相反, AARTiO4(AA=Na、K、Rb)碱金属离子对氧八面体旋转的影响较小, 其作用是施加相邻层的“键应变”.因此, 层间晶格失配解释了A位碱金属尺寸对八面体旋转不稳定性的影响.其SHG信号随温度变化的曲线可分辨出AARTiO4(AA=Na、K、Rb)氧化物属于非中心对称结构, 如图15(c)所示[141].值得注意的是, 稀土离子的配位环境在这些化合物中诱导了氧八面体畸变: 离子半径较小的稀土阳离子与氧离子键合较弱, 驱动氧八面体较大畸变; 而离子半径较大的稀土阳离子键合过强, 氧八面体畸变发生的驱动力较弱.通过调节A位碱金属离子尺寸可实现氧八面体畸变幅度和宏观铁电性的可控调节[141].此外,当没有氧八面体畸变时, 质子层会发生层间滑移, 从而优化了氢键在晶体结构中的键长和键方向[140]. ... Phase transformations during HLnTiO4 (Ln=La, Nd) thermolysis and photocatalytic activity of obtained compounds 2 2015 ... 前人的理论计算表明, R-P型层状钙钛矿材料中, 若A位离子呈有序排列, 则其铁电性有可能增强[26].更为重要的是, 在单层或三层等奇数层R-P材料中也可能出现铁电性, 而在A位离子无序状态下, 这是难以实现的(当n为奇数时, 材料具有n+1偶数层的A位离子, 在完全无序的情况下, 其反铁畸变位移将被完全抵消), 比如具有Pbca空间群的Ca4Ti3O10就属于非极性相[139-142].因此, 研究A位离子有序的R-P材料能有效地拓宽HIF的研究范围.同时, 由于对称性在奇数和偶数层R-P材料中有所差异, 其铁电性起源也可能不同, 因此研究奇数层R-P材料有助于加深对HIF物理本质的理解.2004年, Rodgers等[143]首先利用中子和电子衍射发现了A、B位离子同时有序的单层R-P材料La2Sr2LiRuO8, 其晶体结构如图15(a)所示, 并确认其室温结构为极性Imm2正交相, 但其宏观铁电极化尚未报道.Akamatsu课题组[139-142]借助同步辐射XRD和DFT计算, 研究了A位离子有序单层R-P材料MRTiO4(M=H、Li、Na、K, R为稀土)的晶体结构, 发现氧八面体的倾转破坏了材料的反演对称性, 仅形成了非中心对称的非极性P$\bar{4}$21m四方相.LiRTiO4和NaRTiO4化合物的非中心对称起源于氧八面体的a?b0c0/b0a?c0旋转, 其中LiRTiO4中形成了LiO反萤石层, 而NaRTiO4中形成的是NaO岩盐层(图15(b)), 并且氧八面体畸变幅度的大小取决于稀土阳离子的离子半径[139,142].HRTiO4是单层R-P结构系列的衍生物, 其中TiO6八面体层之间的HO层在结构和化学环境上与HRTiO4[139]和NaRTiO4[142]中的LiO和NaO层完全不同.早期文献指出HRTiO4(R=La、Nd)属于P4/nmm空间群[144], HYTiO4属于P21/c空间群[145], HSmTiO4属于I4/mmm空间群[146].此外, Silyukov等[147]发现HRTiO4(R=La、Nd)氧化物在298~1273 K之间经历了结构相变和脱水.HRTiO4氧化物具有中心对称结构, 可以作为压电材料应用于光催化领域.实际上, 优化稀土离子配位环境是氧八面体旋转的主要驱动力[147].相反, AARTiO4(AA=Na、K、Rb)碱金属离子对氧八面体旋转的影响较小, 其作用是施加相邻层的“键应变”.因此, 层间晶格失配解释了A位碱金属尺寸对八面体旋转不稳定性的影响.其SHG信号随温度变化的曲线可分辨出AARTiO4(AA=Na、K、Rb)氧化物属于非中心对称结构, 如图15(c)所示[141].值得注意的是, 稀土离子的配位环境在这些化合物中诱导了氧八面体畸变: 离子半径较小的稀土阳离子与氧离子键合较弱, 驱动氧八面体较大畸变; 而离子半径较大的稀土阳离子键合过强, 氧八面体畸变发生的驱动力较弱.通过调节A位碱金属离子尺寸可实现氧八面体畸变幅度和宏观铁电性的可控调节[141].此外,当没有氧八面体畸变时, 质子层会发生层间滑移, 从而优化了氢键在晶体结构中的键长和键方向[140]. ...
R-P层状钙钛矿氧化物中的氧八面体旋转和倾侧两种非极性模的耦合可诱导出HIF, 其铁电极化源于A位阳离子的未抵消反铁畸变位移.HIF可以避免常规铁电性和铁磁性的电子构型不兼容的问题,为研究具有强磁电耦合的单相多铁氧化物提供了可能.目前, 基于R-P氧化物的HIF研究工作集中在双层R-P氧化物, 如双层R-P结构的Ca 3M 2O 7(M= Ti、Mn)、Sr 3M 2O 7(M=Sn、Zr)以及Li 2AM 2O 7(A=Ca、Sr; M=Ta、Nb)氧化物.同时, 双层R-P结构的(1? x)(Sr 0.4Ca 0.6) 1.15Tb 1.85Fe 2O 7- xCa 3Ti 2O 7(0.13< x<0.20)固溶体在室温下展现出磁电耦合的单相多铁性.最近, 在A位阳离子有序三层R-P结构的Li 2R 2Ti 3O 10(R= La、Nd)中观测到了室温铁电性, 打破了HIF仅存在于双层R-P结构化合物中的传统观念.目前为止, 报道的单相多铁性氧化物很少, 仅有铁氧体固溶体(1? x)(Sr 0.4Ca 0.6) 1.15Tb 1.85Fe 2O 7- xCa 3Ti 2O 7和Ca 3Mn 2O 7基氧化物两种, 而且铁氧体固溶体的可翻转铁电极化尚需更多直观数据支持.Ca 3Mn 2O 7基氧化物则由于较高的漏电流及中间相Acaa诱导的不规则畴结构, 阻碍了铁电翻转, 导致铁电性难以表征, 后续研究需要进一步降低其漏导电流, 并通过化学压调控中间相来实现铁电翻转.值得关注的是, 最近在La 2SrSc 2O 7基氧化物中发现了HIF, 为探索单相多铁性提供了一种新的思路. ... Hybrid improper ferroelectricity and phase transition behavior of Li2Nd2Ti3O10 ceramics with A-site ordered triple-layer Ruddlesden-Popper structure 7 2024 ... 尽管理论上已经预测并成功合成了许多具有单层R-P结构的非中心对称结构氧化物, 并对部分单层R-P氧化物的压电特性进行了表征, 但其可翻转的铁电极化鲜有报道[139-140,148].本课题组通过放电等离子烧结工艺成功制备了致密的Li2La2Ti3O10陶瓷, 并利用PUND法在室温下观测到了其饱和电滞回线, 测得剩余极化强度为0.25 μC/cm2, 矫顽场为260 kV/cm[149], 如图16(a)所示.结合NPD, 同步辐射XRD和选区电子衍射(SAED)确定Li2La2Ti3O10陶瓷具有极性P21ab空间群的三层R-P结构[149].此外, DFT计算结果显示Pc、P21ab、P21am(No. 26)、A21am和P21an空间群与Li2La2Ti3O10结构精修的拟合优度χ2分别为6.15、4.82、5.12、4.95、5.43, 加权图形剩余方差因子Rwp分别为9.19%、8.16%、8.41%、8.30%、8.66%, 进一步验证了Li2La2Ti3O10氧化物形成能最低的空间群为P21ab(图16(b)).这项工作首次在实验上证实了A位阳离子有序的三层R-P结构的Li2La2Ti3O10陶瓷在室温下具有铁电性, 极大地扩展了HIF的研究领域[149].随后, 本课题组基于NPD数据精修确定Li2Nd2Ti3O10陶瓷在室温下属于极性P21ab相[150].PUND法测得其饱和室温电滞回线, 具有约0.4 μC/cm2的剩余极化强度和150 kV/cm的矫顽场, 如图16(c)所示.Li2Nd2Ti3O10陶瓷的铁电性同样起源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的协同作用[150].与Li2La2Ti3O10陶瓷相比, Li2Nd2Ti3O10陶瓷表现出更高的铁电极化强度和较低的矫顽场, 这可能是源于Nd3+离子半径小于La3+导致更显著的氧八面体畸变(图16(d)), 进而获得了更优异的铁电性能.而较低的矫顽场则与Li2La2Ti3O10陶瓷含有少量的第二相有关.Li2La2Ti3O10陶瓷中的铁电性源于Jahn-Teller畸变、氧八面体畸变和A位阳离子有序的耦合, 其中A位阳离子有序是奇数层R-P结构材料获得极性相的关键因素[149], 其晶体结构如图16(e)所示.图16(f)展示了R-P层状结构中各层极化对宏观极化的贡献, 宏观极化主要源于TiO6氧八面体畸变, 这与双层R-P氧化物的铁电起源完全不同; 双层R-P氧化物的宏观极化是由A位阳离子的未抵消反铁畸变位移引起.A位离子有序三层R-P结构陶瓷的实验工作为进一步拓宽HIF的研究宽度与广度提供了新的见解, 且研究思路可以拓展到其余层状钙钛矿氧化物[149-150]. ...
本文的其它图/表
-
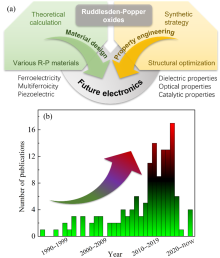 图1
(a)基于R-P结构的HIF的多功能特性总结; (b)基于Web of Science数据库总结的近年发表的R-P氧化物文章数量
图1
(a)基于R-P结构的HIF的多功能特性总结; (b)基于Web of Science数据库总结的近年发表的R-P氧化物文章数量
-
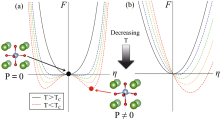 图2
(a)常规铁电相变和(b)非常规铁电相变的能量变化[23]
图2
(a)常规铁电相变和(b)非常规铁电相变的能量变化[23]
-
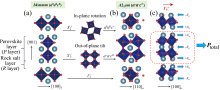 图3
R-P结构A′2AB2O7从(a)顺电相到(b)铁电相的对称模分解及(c)铁电相中每层沿a轴的反铁畸变位移(X)及总的铁电极化强度(Ptotal)示意图[25]
图3
R-P结构A′2AB2O7从(a)顺电相到(b)铁电相的对称模分解及(c)铁电相中每层沿a轴的反铁畸变位移(X)及总的铁电极化强度(Ptotal)示意图[25]
-
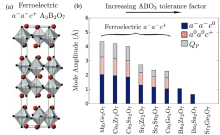 图4
(a) A3B2O7 R-P型氧化物的铁电a−a−c+结构; (b) A3B2O7的氧八面体倾转的第一性原理振幅和诱导极性模式(按ABO3结构τ递增排列)[26]
图4
(a) A3B2O7 R-P型氧化物的铁电a−a−c+结构; (b) A3B2O7的氧八面体倾转的第一性原理振幅和诱导极性模式(按ABO3结构τ递增排列)[26]
-
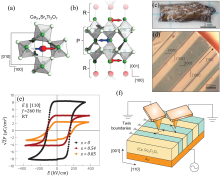 图5
(a, b) (Ca,Sr)3Ti2O7单晶的a0a0c+和a−a−c0两个氧八面体倾转模式的示意图; (c, d) Ca2.46Sr0.54Ti2O7单晶的(c) (001)解理表面照片和(d)室温环形差分干涉衬度照片; (e) Ca3−xSrxTi2O7(x=0, 0.54, 0.85)单晶沿[110]方向的电滞回线; (f) IP-PFM的配置示意图[49]
图5
(a, b) (Ca,Sr)3Ti2O7单晶的a0a0c+和a−a−c0两个氧八面体倾转模式的示意图; (c, d) Ca2.46Sr0.54Ti2O7单晶的(c) (001)解理表面照片和(d)室温环形差分干涉衬度照片; (e) Ca3−xSrxTi2O7(x=0, 0.54, 0.85)单晶沿[110]方向的电滞回线; (f) IP-PFM的配置示意图[49]
-
 表1
双层R-P结构Ca3Ti2O7基氧化物的铁电性能相关参数[43-44,49,64,70 -73,79 -81]
表1
双层R-P结构Ca3Ti2O7基氧化物的铁电性能相关参数[43-44,49,64,70 -73,79 -81]
-
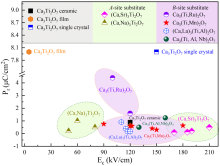 图6
双层R-P结构Ca3Ti2O7基化合物的铁电性能对比[40]
图6
双层R-P结构Ca3Ti2O7基化合物的铁电性能对比[40]
-
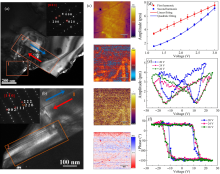 图7
(a)室温下利用g=100衍射斑点得到的DF-TEM照片, 红蓝箭头代表沿[100]的铁电极化方向; (b)利用g=220衍射斑点得到的DF-TEM照片; (c) Ca3[Mn0.5(Fe0.5Nb0.5)0.5]2O7陶瓷室温下的PFM测试结果; (d)改变交流电压条件下的一次与二次谐波压电响应图; (e, f)不同直流偏压下的(e)振幅蝴蝶曲线及(f)相位迟滞回线[45]
图7
(a)室温下利用g=100衍射斑点得到的DF-TEM照片, 红蓝箭头代表沿[100]的铁电极化方向; (b)利用g=220衍射斑点得到的DF-TEM照片; (c) Ca3[Mn0.5(Fe0.5Nb0.5)0.5]2O7陶瓷室温下的PFM测试结果; (d)改变交流电压条件下的一次与二次谐波压电响应图; (e, f)不同直流偏压下的(e)振幅蝴蝶曲线及(f)相位迟滞回线[45]
-
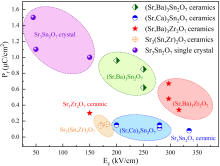 图8
双层R-P结构Sr基氧化物(Sr3Sn2O7单晶[112]、(Sr,Ba)3Sn2O7陶瓷[84]、(Sr,Ca)3Sn2O7陶瓷[85]、(Sr,Ba)3Zr2O7陶瓷[86]、Sr3(Sn,Zr)2O7陶瓷[113])的铁电性能对比
图8
双层R-P结构Sr基氧化物(Sr3Sn2O7单晶[112]、(Sr,Ba)3Sn2O7陶瓷[84]、(Sr,Ca)3Sn2O7陶瓷[85]、(Sr,Ba)3Zr2O7陶瓷[86]、Sr3(Sn,Zr)2O7陶瓷[113])的铁电性能对比
-
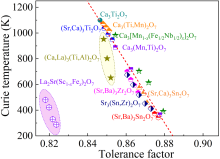 图10
双层R-P结构的铁电体的TC与τ的关系[40]
图10
双层R-P结构的铁电体的TC与τ的关系[40]
-
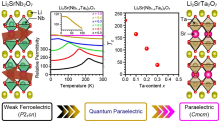 图11
Li2Sr(Nb1−xTax)2O7氧化物的介电常数与温度的依赖关系以及铁电相变模型[65]
图11
Li2Sr(Nb1−xTax)2O7氧化物的介电常数与温度的依赖关系以及铁电相变模型[65]
-
 图12
(a) La2SrSc2O7陶瓷的晶体结构示意图, 室温下的P−E电滞回线和不同A位离子有序度下的DFT计算能量对比[123]; (b) La2Sr(Sc1−xFex)2O7陶瓷在400 kV/cm电场和2 Hz频率下的P−E曲线[121]; (c) La2Sr(Sc1−xFex)2O7陶瓷在150~600 K温度范围内的介电常数的温度依赖性[121]; (d) La2Sr(Sc1−xFex)2O7(x=0.15)陶瓷直流磁化率的温度依赖性, 插图是Curie−Weiss拟合结果[121]; (e) La2Sr(Sc1−xFex)2O7(x=0.15)在不同温度下磁化的等温磁场依赖性[121]
图12
(a) La2SrSc2O7陶瓷的晶体结构示意图, 室温下的P−E电滞回线和不同A位离子有序度下的DFT计算能量对比[123]; (b) La2Sr(Sc1−xFex)2O7陶瓷在400 kV/cm电场和2 Hz频率下的P−E曲线[121]; (c) La2Sr(Sc1−xFex)2O7陶瓷在150~600 K温度范围内的介电常数的温度依赖性[121]; (d) La2Sr(Sc1−xFex)2O7(x=0.15)陶瓷直流磁化率的温度依赖性, 插图是Curie−Weiss拟合结果[121]; (e) La2Sr(Sc1−xFex)2O7(x=0.15)在不同温度下磁化的等温磁场依赖性[121]
-
 图13
双层R-P材料(Ca3Ti2O7陶瓷[80]、Sr3Sn2O7陶瓷[84]、Sr3Zr2O7陶瓷[86]、Li2CaTa2O7陶瓷[26]、Li2SrNb2O7陶瓷[130])铁电性能的对比
图13
双层R-P材料(Ca3Ti2O7陶瓷[80]、Sr3Sn2O7陶瓷[84]、Sr3Zr2O7陶瓷[86]、Li2CaTa2O7陶瓷[26]、Li2SrNb2O7陶瓷[130])铁电性能的对比
-
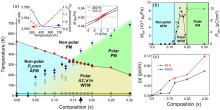 图14
(a) (1−x)(CaySr1−y)1.15Tb1.85Fe2O7-xCa3Ti2O7(0≤x≤0.3, y=0.60)的相图; (b)铁电极化与饱和磁化强度随成分的变化; (c)当温度为60和100 K时, 线性磁电耦合系数随成分的变化[136]
图14
(a) (1−x)(CaySr1−y)1.15Tb1.85Fe2O7-xCa3Ti2O7(0≤x≤0.3, y=0.60)的相图; (b)铁电极化与饱和磁化强度随成分的变化; (c)当温度为60和100 K时, 线性磁电耦合系数随成分的变化[136]
-
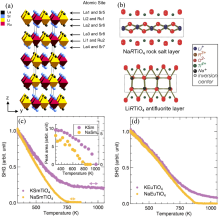 图15
(a)单层R-P结构氧化物的晶体结构[143]; (b) HRTiO4和NaRTiO4的插层结构示意图[139]; (c) AASmTiO4和(d) AAEuTiO4的SHG信号随温度升高的变化曲线, 其中AA为Na(黄色圆圈)和K(紫色圆圈)[141]
图15
(a)单层R-P结构氧化物的晶体结构[143]; (b) HRTiO4和NaRTiO4的插层结构示意图[139]; (c) AASmTiO4和(d) AAEuTiO4的SHG信号随温度升高的变化曲线, 其中AA为Na(黄色圆圈)和K(紫色圆圈)[141]
-
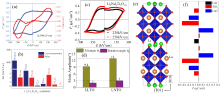 图16
(a) 390 kV/cm条件下采用PUND法测得的Li2La2Ti3O10陶瓷的P−E电滞回线和J−E电流密度曲线[149]; (b)基于DFT计算的三层R-P结构Li2La2Ti3O10的不同对称性相对于0 K时能量最低Pc相的能量[149]; (c) PUND法测得的Li2Nd2Ti3O10陶瓷的室温P−E电滞回线[150]; (d) Li2La2Ti3O10(LLTO)和Li2Nd2Ti3O10(LNTO)陶瓷的氧八面体畸变角度θT和θR[150]; (e)基于Rietveld精修的Li2La2Ti3O10陶瓷晶体结构示意图(绿色、棕色和红色球分别代表Li+、La3+和O2−离子, 而Ti4+离子位于氧八面体的中心)[149]; (f)通过玻恩有效电荷方法计算的Li2La2Ti3O10中每一层对应的极化贡献[149]
图16
(a) 390 kV/cm条件下采用PUND法测得的Li2La2Ti3O10陶瓷的P−E电滞回线和J−E电流密度曲线[149]; (b)基于DFT计算的三层R-P结构Li2La2Ti3O10的不同对称性相对于0 K时能量最低Pc相的能量[149]; (c) PUND法测得的Li2Nd2Ti3O10陶瓷的室温P−E电滞回线[150]; (d) Li2La2Ti3O10(LLTO)和Li2Nd2Ti3O10(LNTO)陶瓷的氧八面体畸变角度θT和θR[150]; (e)基于Rietveld精修的Li2La2Ti3O10陶瓷晶体结构示意图(绿色、棕色和红色球分别代表Li+、La3+和O2−离子, 而Ti4+离子位于氧八面体的中心)[149]; (f)通过玻恩有效电荷方法计算的Li2La2Ti3O10中每一层对应的极化贡献[149]
|
{kind=link}