Pt3Co高指数晶面氧还原过程的密度泛函理论研究
刘磊, 郭瑞华, 王丽, 王艳, 张国芳, 关丽丽
无机材料学报
2025, 40 ( 1):
39-46.
DOI:10.15541/jim20240296
Pt3Co催化剂是Pt基合金中氧还原反应(ORR)活性最高的催化剂, 合成Pt3Co高指数晶面(HIFs)是一种提高其催化性能的有效策略, 但拥有最高ORR活性的HIFs尚未明确, 并且目前缺乏对Pt3Co HIFs ORR的系统研究。本研究构建了六种不同Pt3Co HIFs, 通过从头算分子动力学(AIMD)计算证明了其稳定性, 通过密度泛函理论(DFT)计算了六种Pt3Co HIFs的ORR过程中间物*O、*OH、*OOH的结合能(BE), 通过d带中心(εd)、Bader电荷及配位数(CN)解释了其在台阶与边缘位点BE不同的原因。同时分析了吸附原子CN与εd的关系, 通过ORR自由能台阶图分析了ORR过程中的过电位(η), 发现η大小主要与*OH结合能(BE-*OH)有关, 其中η最小的晶面为Pt3Co(211), 其在台阶处的η达到了0.294 eV。本工作为高ORR活性HIFs催化剂研发提供了一定的理论依据。
| HIFs | Microfact notation | k-point | Number of atoms | | Pt3Co(211) | n(111)×(100) | 3×2×1 | 48 | | Pt3Co(310) | n(100)×(110) | 3×2×1 | 48 | | Pt3Co(331) | n(110)×(111) | 2×2×1 | 48 | | Pt3Co(511) | n(100)×(111) | 2×2×1 | 48 | | Pt3Co(320) | n(110)×(100) | 3×2×1 | 48 | | Pt3Co(332) | n(111)×(110) | 2×2×1 | 48 |
View table in article
表S1
六个HIFs的微面标记方式、k点、原子数
正文中引用本图/表的段落
采用基于DFT的VASP[22]软件进行计算, 其中Perdew-Burke-Ernzerhof(PBE)[23]为交换关联能量泛函, 所有计算采用了Grimme等[24]提出的D3色散校正。采用投影缀加波(PAW)方法解决Kohn-Sham方程[25], 截断能设置为400 eV的平面波基组[26]。不同晶面对应的布里渊区(BZ)积分的k点网格设置如补充材料表S1所示。计算过程中考虑了自旋极化, 为了获得平衡构型, 每个原子上的力小于0.5 eV/nm, 且总能量收敛为10-5 eV[27]。在晶格的z方向设置了1.5 nm的真空层[28], 以避免周期图像之间不必要的相互作用。此外, 本研究还采用从头算分子动力学(AIMD)计算表面模型, 以1 fs的时间步长评估系统在300 K下2 ps的热力学稳定性[29]。
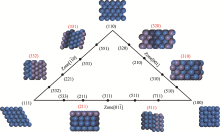
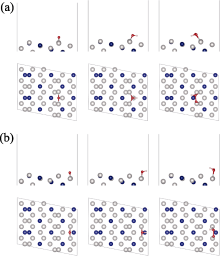
图1为不同晶面的示意图。对于面心立方金属, 图1中的立体三角形代表不同晶面对应的位置, 三角形中的三个顶点表示三个低指数晶面, 即(100)、(110)和(111)。三条边线分别代表[1$\overline{1}$0]、[001]、[01$\overline{1}$]晶带, 三角形边线上的位置则表示不同HIFs[34]。在这三条晶带中, 不同HIFs含有不同类型的阶梯位点。在[1$\overline{1}$0]、[001]、[01 $\overline{1}$]晶带中, 一共存在三种阶梯位点, 即(331)、(332), (310)、(320)和(211)、(511)。根据微面标记法, 其一般形式为n(htktlt)×(hsksls), 它表示n个原子宽度的(htktlt)梯田被一个(hsksls)阶梯所分开。则(331)、(332)可以表示为n(110)×(111)、n(111)×(110), (310)、(320)可以表示为n(100)×(110)、n(110)×(100), (211)、(511)可以表示为n(111)×(100)、n(100)×(111)[35-36]。因此, 本研究选取的Pt3Co HIFs主要为Pt3Co(211)、Pt3Co(511)、Pt3Co(310)、Pt3Co(320)、Pt3Co(331)、Pt3Co(332)[37]。目前, (211)[38]、(332)[39]HIFs可以通过水热法合成, (511)[40]、(310)[41]HIFs可以通过电化学法合成, (320)[42]HIFs可以通过三次分区精炼法合成, (331)[43]HIFs可以通过种子介导生长法合成。
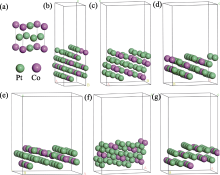
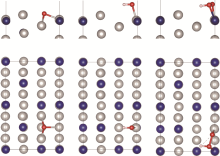
首先对Pt3Co原胞进行优化, 优化后, Pt3Co晶胞的晶格常数为0.383 nm, 与文献[44]相吻合(Pt3Co: 0.388 nm)。然后对Pt3Co晶胞进行切面、扩胞, 保持Pt、Co原子比为3:1, 再进行结构优化, 得到六个吸附表面模型, 如图2所示。为了评估其稳定性, 计算了六个表面的表面能。结果表明Pt3Co(211)、Pt3Co(310)、Pt3Co(331)、Pt3Co(511)、Pt3Co(320)、Pt3Co(332)的表面能分别为1.537、1.921、1.696、1.881、1.820、1.569 J/m2, 与Zeng等[45]的计算结果基本吻合。同时还对表面模型进行了AIMD计算, 结果如图S1所示。在T=300 K时, Pt3Co(211)、Pt3Co(310)、Pt3Co(331)、Pt3Co(511)、Pt3Co(320)、Pt3Co(332)的表面能量趋于稳定, 证明所构建的六个表面具有良好的热力学稳定性。
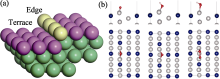
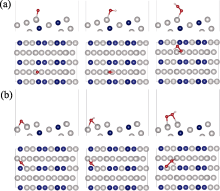
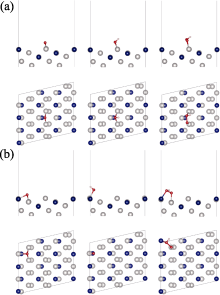
本研究计算了ORR过程中*O、*OH、*OOH中间物在Pt3Co(211)、Pt3Co(310)、Pt3Co(331)、Pt3Co(511)、Pt3Co(320)、Pt3Co(332) HIFs不同位置上的吸附情况, 其中吸附位点主要分为两类, 分别是台阶(Terrace, 紫色)和边缘(Edge, 黄色)[46-47], 如图3(a)所示。根据两类吸附位点的计算结果各自选取了最佳吸附位点, 其中图3(b)为Pt3Co(211)边缘处的最佳吸附构型, 其余HIFs最佳吸附构型见补充材料图S2~图S7, *O、*OH、*OOH在六个HIFs不同位置的BE及吸附距离(BD)如表1所示。
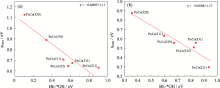
通过表1中不同晶面、不同位置处*O、*OH、*OOH的BE可以看出, 在Pt3Co(211)、Pt3Co(310)、Pt3Co(331)、Pt3Co(511)、Pt3Co(320)、Pt3Co(332)六个晶面台阶处对*O、*OH、*OOH的吸附作用大多数都小于在边缘处的吸附, 且在Pt3Co(211)台阶和边缘处对*O、*OH的吸附作用小于在其他五个HIFs。
本文的其它图/表
|