气凝胶是一类由纳米颗粒构成并呈现三维网络骨架结构的多孔固体材料[1 ] 。由于构成骨架的固体颗粒和孔隙结构均为纳米量级, 因而这一独特结构使气凝胶呈现出如低密度、高孔隙率、高比表面积、低热导率、高透过率和高吸附率等特征和性能, 并可作为光学传感器、隔热材料、隔声材料、催化剂及载体、吸附剂、电极材料和惯性约束聚变靶材等使用, 具有广泛的应用前景[1 -8 ] 。
除了单组份气凝胶材料以外, 双元或多元气凝胶则是结合不同组分间的优点形成具有新特性的复合气凝胶材料。其中, C/SiO2 复合气凝胶材料是研究较多并具有丰富复合结构形式的一类材料。根据应用背景的不同, 该复合材料可在保持某一组元特性的同时, 通过引入另一组元提高单一组元材料的性能。比如, 在SiO2 气凝胶中引入碳源作为红外遮光剂, 可以提高SiO2 气凝胶的抗辐射能力[9 ] ; 在活性炭中引入SiO2 气凝胶可以提高材料的吸附率[10 ] 等等。若对C/SiO2 复合气凝胶材料进行高温烧结, 经碳热还原(carbothermal reduction)反应, 可转化为C/SiC复合材料[11 ] 或SiC单体材料[12 ] 。基于此方法获得的具有气凝胶结构特征的SiC材料, 除保持SiC本身的高硬度、高抗热震能力、高热导率、高热稳定性等特点外, 还具有高孔隙率、高比表面积等多孔材料的特征, 因而可在结构陶瓷、催化剂载体和过滤器等领域得以应用[12 -13 ] 。
通常, 基于溶胶-凝胶法制备的C/SiO2 和C/SiC复合材料具有气凝胶材料的结构特征。碳化前, 借助于均匀分布的单元或双元溶胶胶体颗粒实现硅元素与碳元素的复合。一般而言, SiO2 常以有机硅氧烷为先驱体经水解缩聚后形成的SiO2 溶胶方式引入。而对于碳源的引入, 由于碳材料的多样性, 如催化丝状碳(catalytic filamentous carbon, CFC)、Sibunit碳、颗粒状碳黑(granular carbon black)、活性炭(activated carbon)、碳编织体(carbon weave)、碳毡(carbon felt)[14 ] 以及由有机聚合物提供的碳源[15 ] 等, 碳的引入呈现出复杂的形式。根据硅源与碳源引入方式的不同, 本文将基于溶胶-凝胶法制备的具有气凝胶材料结构特征的C/SiO2 和C/SiC复合材料的制备方式主要分为三类, 即共聚法(co-polymerization)、浸入法(impregnation)和聚合物先驱体热解法(polymeric precursors pyrolysis)。
1 共聚法
共聚法是指硅源和碳源分别以SiO2 溶胶和有机溶胶的形式引入, 经均匀混合、凝胶和干燥后, 形成有机-SiO2 气凝胶, 再经高温烧结获得C/SiO2 和C/SiC复合材料。该方法通常采用有机硅氧烷醇盐为先驱体配制SiO2 溶胶, 而碳源通常以间苯二酚(resorcinol, R)和甲醛(formaldehyde, F)聚合物先驱体的形式引入, 经水解和缩聚后形成间苯二酚-甲醛(resorcinol-formaldehyde, RF)溶胶。凝胶后, 形成的混合凝胶中有机骨架与SiO2 骨架相互交叉缠绕, 但通常这两种网络骨架之间并无化学键连接。由于RF气凝胶的溶胶-凝胶过程相较于SiO2 十分缓慢[16 -18 ] , 因而RF与SiO2 凝胶形成时间的不同往往会在SiO2 凝胶骨架表面形成RF凝胶。为了更好地获得均匀的RF-SiO2 气凝胶, 通常采用分别溶胶再混合凝胶的方法制备RF-SiO2 气凝胶。图1 给出了RF-SiO2 气凝胶的形成过程及其向SiC气凝胶的转变示意图[12 ] 。
图1 RF-SiO2 气凝胶的形成过程及其向SiC气凝胶的转变示意图[12 ]
Fig. 1 Formation of RF-SiO2 aerogel and its transformation to SiC aerogel[12 ]
基于上述考虑, 当采用正硅酸乙酯(tetraethoxysilane, TEOS)为先驱体水解得到SiO2 溶胶, 加入到事先配制的RF溶胶中, 可以获得均匀的混合溶胶。混合溶胶经凝胶、干燥后获得RF/SiO2 气凝胶, 随后在惰性气体保护下热解得到C/SiO2 气凝胶材料[11 -12 ,19 -20 ] 。TEOS加入RF溶胶能明显缩短体系的凝胶时间, 但是随着TEOS含量的逐渐增加, 气凝胶孔径逐渐减小, 孔内毛细张力增大, 气凝胶易于开裂[21 ] 。Chen等[22 -23 ] 针对该混合溶胶体系, 采用一步酸催化快速合成法(one-pot acid-catalyzed rapid synthesis route)制备了RF/SiO2 气凝胶。在该方法中, HCl取代Na2 CO3 作为酸性催化剂, 可以提高芳香族链亲电取代(electrophilic aromatic substitution)的活性, 从而加速芳香族链之间亚甲基桥联的形成; 乙腈(CH3 CN)取代乙醇作为溶剂, 为TEOS的溶胶-凝胶过程提供了更加合适的环境, 适合于一步形成RF/ SiO2 体系。这种在CH3 CN溶剂中采用酸性催化剂的反应体系可以显著加快RF先驱体的反应速率, 可以将混合体系的凝胶时间由几天缩短至几个小时。RF/SiO2 复合气凝胶材料的孔结构呈现与纯SiO2 和纯RF气凝胶相似的结构特征, 并具有低的热导率和高的力学行为。图2 给出了酸催化RF/SiO2 凝胶的形成过程[22 ] 。
为了避免以TEOS或正硅酸甲酯(tetramethy lorthosilicate, TMOS)为硅源制备RF/SiO2 气凝胶时的复杂溶胶-凝胶过程, 进一步减少水解缩聚的时间, 有研究采用硅烷偶联剂作为硅源一步合成RF/ SiO2 气凝胶[15 ,24 -26 ] 。在该类方法中, 硅烷偶联剂3-氨基丙基三乙氧基硅烷[(3-aminopropyl)triethoxysilane, APTES]可作为硅源, 直接与R和F混合形成溶胶, 凝胶并超临界干燥后获得RF/SiO2 复合气凝胶[15 ,24 -25 ] 。APTES含量的增加将减少混合溶胶体系的凝胶时间[24 ,27 ] 。在该溶胶体系中, APTES除了作为硅源先驱体参与水解缩聚反应形成Si-O-Si网络结构以外, 还可作为“内部催化剂(internal catalyst)”促进R和F之间的缩聚反应。这是由于APTES中氨基的水合作用(hydration of amino groups)在APTES水解产物中形成了环状的分子间氢键, 为反应提供了良好的通路, 从而加速溶胶-凝胶过程[27 ] 。RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] 。以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度。经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] 。APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] 。除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] 。
图2 酸催化RF/SiO2 凝胶的形成过程示意图[22 ]
Fig. 2 Formation of acidcatalyzed RF/SiO2 hybrid gels[22 ]
当热解温度提高至1200~1500℃, RF/SiO2 气凝胶或C/SiO2 气凝胶经碳热还原反应转变为SiC或SiC/C气凝胶, 总的反应方程如式(1)所示[12 ,20 ] :
SiO2 (s)+3C(s)→SiC(s)+2CO(g) (1)
在这一过程中, SiC首先在先驱体表面形核(nuclei), 随后长大成纳米晶须(nanowhiskers)。其间相互贯穿孔的存在为气体产物(如SiO、CO和CO2 )的扩散提供了通路, 加速了这一反应的进行[12 ] 。C与SiO2 的比例和C/SiO2 复合材料的致密度是影响SiC反应动力学和最终SiC微观结构的重要因素。图3 给出了不同结构SiC的形成过程示意图[28 ] 。高碳硅比例下, 介孔C/SiO2 可形成纳米级纤维状和颗粒状SiC, 如图3 (a)所示; 低碳硅比例下, 除形成纳米纤维状和颗粒状SiC以外, 还渗透着少量自由C, 如图3 (b)所示。其中, 纤维状和颗粒状SiC产物的比例可通过改变碳硅间的比例并利用C的渗透加以调节。高碳硅比例的致密C/SiO2 可转变为介孔SiC, 如图3 (c)所示。这是由于高碳硅比例以及C/SiO2 和C之间界面结构可以提高碳热还原反应速率, 介孔在为气态产物提供扩散通道的同时, 也会限制介孔C/SiO2 材料中SiC的生长[28 ] 。除此以外, 采用低温镁热反应(low-temperature magnesiothermic reaction)可使C/SiO2 材料在700℃的低温下转变成SiC[23 ] 。通过碳热还原RF/SiO2 气凝胶制得的材料是以β- SiC为主体, 并含有少量α-SiC的SiC或C/SiC气凝胶材料。该材料具有多孔结构和高的比表面积, 并具有高硬度、好的抗热震能力、高热导和高稳定性、低膨胀系数和大的带隙, 可用于增强相、催化剂、高能和高频电子材料、光电材料、抗辐射材料、氢分离膜载体和吸波器件等[12 ] 。
图3 SiC形成过程示意图[28 ]
Fig. 3 Schematic illustration of SiC formation[28 ]
采用共聚法制备C/SiO2 和C/SiC气凝胶, 除了以R和F的聚合物为先驱体之外, 碳的来源还可能是间苯三酚-F(Phloroglucinol-Formaldehyde, PF)[29 ] 、可溶性酚醛树脂(resol)[30 ] 、苯酚-R-F(phenol-resorcinol- formaldehyde, PRF)[31 ] 、蔗糖(saccharose)[32 ] 、淀粉(starch)[33 ] 、石油焦(petroleum coke)[34 ] 、煤沥青(coal tar pitch)[35 ] 、酚醛清漆(novolac)[36 ] 等。从经济角度来看, 以经济衍生物作为碳源可以有效地改善R作为原料时价格高的缺点, 但这种碳源成分较复杂。而以蔗糖、淀粉、石油焦等作为碳源很难能形成交联的网络结构, 无法得到高孔隙率的块状气凝胶材料。
2 浸入法
浸入法是指将碳材料作为模板或增强相浸入SiO2 溶胶形成C/SiO2 复合材料的方法。根据该类材料的不同应用背景, 所用碳材料可选择碳粉、活性炭、碳泡沫、碳气凝胶和碳毡等多种形式。正是由于碳材料存在的多样性, 引起碳与SiO2 之间多样的复合存在形式。常见的复合形式主要有: (1)将SiO2 溶胶浸入以碳材料构成的多孔骨架结构中, SiO2 以填充孔隙的形式存在; (2)将碳颗粒引入SiO2 溶胶中, 待凝胶和干燥后, 实现碳颗粒对SiO2 基体材料的增强和改性; (3)SiO2 溶胶包裹碳材料, 实现对碳材料的改性。碳与SiO2 之间这种多样的复合形式, 使C/SiO2 复合材料在电池、储氢、催化、吸附、隔热等领域具有广泛的应用前景。
在吸附领域, 活性炭经SiO2 溶胶改性后, 借助于SiO2 气凝胶纳米多孔的结构特性, 可以提高活性炭材料的比表面积和孔隙率, 进而提高活性炭的吸附率。Zhou等[10 ] 将活性炭浸入以TEOS和苯基三乙氧基硅烷(phenyltriethoxysilane, PTES)或甲基三乙氧基硅烷(methyltriethoxysilane, MTES)为混合先驱体制备的SiO2 溶胶中, SiO2 溶胶颗粒原位聚合于碳材料表面, 获得了SiO2 /活性炭复合材料。该材料的微观结构上是SiO2 气凝胶包裹活性炭颗粒, 且骨架结构比较松散。依靠表面吸附(surface adsorption)作用, 将其吸附率从活性炭的76%提高到96.5%。该材料可实现从废水中吸附三硝基甲苯(2,4,6-trinitrotoluene, TNT)的作用。同时, 有机硅氧烷先驱体提供的不同疏水基团对吸附率也有影响。Karnib等[37 ] 用APTES对SiO2 颗粒进行表面修饰后, 与活性炭在溶液状态下混合, 经干燥和研磨后, 获得的SiO2 /活性炭复合颗粒对水溶液中的重金属离子具有很好的吸附效果。
在隔热领域, 由于碳材料对红外波段具有良好的吸收特性, 通常引入SiO2 气凝胶提高其高温抗辐射能力。Lu等[38 ] 将炭黑作为红外遮光剂分散在SiO2 气凝胶中, 可以明显降低高温辐射热传导。Liu等[39 ] 将碳泡沫浸入SiO2 溶胶中, 凝胶并干燥后形成了碳泡沫/ SiO2 气凝胶复合材料。由于碳泡沫表面被SiO2 气凝胶覆盖形成核壳结构, 该复合材料的热导率比纯碳泡沫降低了41.9 %, 但其压缩强度并没有明显下降。
在电催化方面, 经SiO2 修饰的碳载铂催化剂(carbon-supported platinum catalysts), 其热稳定性和电催化特性得以提高, 可用于低温质子交换膜(proton exchange membrane, PEM)燃料电池的电催化领域[40 ] 。
除此以外, 在SiO2 气凝胶中填充炭粉, 可增强材料的弹性回复能力, 并保持SiO2 气凝胶基体的硬度, 这有助于减小气凝胶在超临界干燥过程中的收缩[41 ] , 并可获得更高比表面积的C/SiO2 复合材料[42 ] 。C和SiO2 在C/SiO2 复合材料中的相对分布状态与各自在溶液中的等电点(isoelectric points, IEPs)有关[43 ] 。Spassova等[43 ] 研究发现, 与SiO2 进行复合时, 如果不同形态碳材料的IEPs高于SiO2 的IEPs(pH=2.5), 则基于静电反应(electrostatic interaction)形成以SiO2 为核、C为壳层的结构形式; 如果碳材料的IEPs低于SiO2 的IEPs, 则将出现C颗粒表面吸附Si-OH的结构形式。经SiO2 溶胶浸入后的多孔碳材料(如碳气凝胶), 由于碳骨架表面包裹了一层耐氧化的SiO2 凝胶, 可以提高碳材料的热稳定性[44 ] 。同时, Worsley等[44 ] 发现将比表面积高达3000 m2 /g的碳气凝胶浸入SiO2 溶胶后, 由于纤细的碳骨架表面均匀覆盖着一层SiO2 凝胶, 为C与SiO2 之间的充分化学反应创造了条件。因而, 经1500℃碳热还原反应后, C/SiO2 复合气凝胶材料可转化为比表面积达2000 m2 /g的SiC多孔材料。这种方法为形成高比表面积的金属氧化物、碳化物或氮化物提供了一种好的研究思路。另外, C/SiO2 复合材料在高温下向SiC转变的过程中, 由共聚法获得的C/SiO2 气凝胶转变成SiC时, 常以纳米晶须状(whiskerlike)而不是颗粒状(particulate)的形式存在, 但这一晶须状结构通常并不是理想的SiC材料形式。为了克服这一问题, 通常需要在SiO2 表面包覆C的先驱体, 即形成C包SiO2 的胶囊式结构, 以确保所有在SiO2 和C界面间产生的气态SiO与C发生反应, 进而形成颗粒状SiC材料[45 ] 。图4 给出了SiO2 与C界面间SiC形成的过程示意图[45 ] 。
3 聚合物先驱体热解法
聚合物先驱体热解法是指以溶胶-凝胶法形成的有机硅氧烷聚合物凝胶经高温热解转变为SiCO (silicon oxycarbide)或SiC材料的方法。SiCO具有C和O同时与Si相结合的化学结构特征。这一四面体网络结构可定义为Cx 4- x x =1, 2或3)[46 ] 。相较于O2- 仅可提供两个阴离子配位键而言, C取代O后, 有提供3到4个阴离子配位的可能性, 这使得SiCO比纯SiO2 具有更高的交联度[47 ] 。基于该方法制备的SiCO, 常具有耐高温、轻质、耐腐蚀、低热导等性能特点, 在高温传感器、催化、过滤、电池电极材料以及高温热防护等领域具有广泛的应用前景。
图4 SiO2 与C界面间SiC形成的过程示意图[45 ]
Fig. 4 Processes of formed silicon carbide at the interface between silica and carbon[45 ]
采用溶胶-凝胶法合成SiCO是一种最佳的制备方法。为了在这一过程中均匀地引入Si-C键, 常以带有直接与Si原子相连烃基的有机硅氧烷作为先驱体, 即烃基取代硅氧烷先驱体(alkyl-substituted silicon-alkoxide precursors)[46 ] 。该先驱体的分子式可表示为Rx 4- x x 可取1~3), 其中, R既可为饱和烃基(如CH3 、C2 H5 ), 也可为不饱和烃基(如C6 H5 ); R’主要为CH3 或C2 H5 [48 ] 。在溶胶-凝胶过程中, 与Si直接相连的R并不参与水解, 因而可保留在以Si-O-Si为主体构成的网络结构中。R中含有的碳链长短和数量, 将决定SiCO中Si-C键的数量, 因此, 先驱体的分子组成是影响SiCO最终构成的最直接因素。有研究表明[49 ] , 凝胶中C含量会随着R基团中碳链长度的增加而呈比例增加。但是, 热解后保留的C含量与先驱体或凝胶中存在的碳链长度并无明确比例关系。随着R基团中C含量的降低, C-Si-O键所占比例将增加。热解后, 只有直接与Si原子相连的C原子才可以SiCO的形式保留在材料中, 而其他C原子则以自由C的形式存在于材料中。
通常, 烃基取代硅氧烷先驱体在水解缩聚过程中, 由于不可水解R基团的存在, 往往对硅烷醇的缩聚造成阻碍, 从而使其难以凝胶。因此, 在溶胶-凝胶阶段常借助易于水解缩聚的四烷氧基硅烷(如TMOS、TEOS)构成交联网络结构。Babonneau等[50 ] 采用二甲基二乙氧基硅烷(dimethyldiethoxysilane, DEDMS)和TEOS作为双先驱体, 经水解缩聚后形成了Si-C-O凝胶体系。29 Si核磁共振(nuclear magnetic resonance, NMR)证明这两种双先驱体之间发生了共聚合化作用, 并存在两类凝胶单元, 即源于DEDMS构成的(CH3 )2 Si(O0.5 )2 和源于TEOS构成的Si(O0.5 )4 。Liu等[42 ] 采用苯基三甲氧基硅烷(phenyltrimethoxysilane, PhTMS)和TMOS作为双先驱体制备的凝胶体系由O3/2 -Si-C6 H5 和1/2 O-Si-O1/2 凝胶单元构成。随着PhTMS含量的增加, 孔大小、孔体积和比表面积均下降, 经1000℃热解后, 获得的SiCO仍旧保持 581 m2 /g的高比表面积。Feng等[51 ] 以TEOS和聚二甲基硅氧烷(polydimethylsiloxane, PDMS)为双先驱体, 经溶胶-凝胶、超临界干燥和1200℃热解后得到块状SiCO气凝胶单体。该材料呈非晶态, 其网络结构可在1100℃下保持稳定, 当温度达到1200℃, 非晶态中逐渐开始出现晶相, 而此时仅有1.65%的质量损失, 可见该材料具有很高的热稳定性。另外, 该材料的密度为0.3 g/cm3 , 热导率仅为0.027 W/(m·K), 可作为一种高温隔热材料使用。
为了最大程度的保留Si-C键, 减少自由C的含量, 可选用含有Si-H基团的有机硅氧烷作为先驱体。有机硅氧烷先驱体中含有Si-H基团, 在减少Si-O键数量的同时, 还会引起 Si-H与Si-Cx y 2 -Si≡键合形式的网络聚合结构。Singh等[48 ] 以甲基二甲氧基硅烷(methyldimethoxysilane, MDMS)和TEOS为双先驱体, 在酸性条件下水解和缩聚后, 得到了多孔SiCO凝胶。基于溶解-再沉淀机制(dissolution-reprecipitation mechanism), 经氨水老化处理, 可实现凝胶孔结构的调节。研究表明, 氨水老化处理后, 凝胶中的(CH3 )HSiO2 结构将转变为(CH3 )SiO3 ; 经高温热解后, 相较于未经氨水老化处理的凝胶来说, 结构中存在更少量的CSiO3 和C2 SiO2 。Si-H键的存在将通过与Si-CH3 反应生成≡Si-CH2 -Si≡键合形式, 进而增加SiCO的含量。因此, 在凝胶中需要保留更多的Si-H键。
除此以外, 为了克服硅氧烷聚合物先驱体热解时的高致密倾向, 多孔SiCO气凝胶型材料逐渐成为研究中的热点。该研究目前主要通过选用[52 -58 ] 或合成[59 ] 不同类型的硅氧烷先驱体, 经溶胶-凝胶、干燥以及高温裂解后, 制备具有多孔结构特征的SiCO。表1 总结了以不同硅氧烷先驱体合成SiCO的孔结构参数, 从中可以看到, SiCO的比表面积和孔体积往往随着裂解温度的升高而降低, 密度则随着裂解温度的升高而增大。这与高温裂解造成材料收缩和致密化有关。另外, 有研究表明, 在400~600℃, 由于热分配反应(thermal redistribution reactions)存在气体释放过程, 往往形成新的临时微孔, 进而提高比表面积和孔体积[53 ,58 ] 。温度继续升高后, 新的交联反应引起孔隙收缩, 造成孔结构参数降低[53 ] 。
高温热解后, Si、O和C三种元素间的化学构成和分布是SiCO研究中的重要方面。由于热解过程常伴随着剧烈的化学反应和材料收缩, 获得的SiCO致密性往往较高。在热解过程中, 温度高于300℃时, Si-C键开始断裂, 并逐渐向Si-O键转变。SiO2 网络结构中存在的Si-C键一直可保持到900℃[50 ] 。经800℃~1000℃高温热解后, SiCO主要由具有自由网络形式的Si-O键和Si-C键构成, 其形式为不同比例的SiC4 、SiOC3 、SiO2 C2 、SiO3 C和SiO4 , 并且含有一定量的自由C。这一复合成分常可表示为: SiCx 2(1- x ) +y Cfree [60 -61 ] 。当热解温度高于1000℃~1200℃后, SiCO将发生相变, 形成SiC、C(turbostratic)和SiO2 (cristobalite)三相的平衡。在这一相变过程中, 非晶SiC和C出现在非晶SiO2 或SiCO基体中[60 ] 。热解温度高于1300℃时, SiO2 在碳热还原作用下再次生成Si-C键, 达到1500℃后, 可获得晶态SiC[50 ] 。Bréquel等[61 ] 为了研究不同化学计量比下SiC和SiO2 复合材料的高温晶化行为, 通过改变双先驱体MDES和TrEOS两者间的相对含量, 设计了三种凝胶配比, 即满足SiCO化学计量的配比、富碳配比和富硅配比。研究表明, 高C含量下, SiCO易于转变为β-SiC晶体; 低C含量下, 易于获得大尺寸晶粒。C的存在将阻碍SiC晶核与SiCO相之间的反应, 因而C富集区域内SiCO转变为SiC的速率变缓。除此以外, SiCO中SiO2 相的高温稳定性也能得以提高[61 ] 。造成这一现象的原因可能是SiO2 结构中残余Si-C键可以阻碍SiO2 结构的晶化重组, 并且由于SiO2 团簇的尺寸太小, 难以形成连续的SiO2 基体。通常, 先驱体中与Si直接相连的烃基类型决定了自由C的含量[62 ] 。饱和烃基基团通常形成适量的自由C, 不饱和烃基基团易于形成更高含量的自由C。当C含量足够高, 由于碳热还原反应的作用, 将使SiO2 转变为SiC, 同时释放CO。
经热解法制备的SiCO比SiO2 具有更高的力学、热学和化学稳定性[46 ] 。在SiCO中, C以自由碳或以与Si连接形成SiC4- x x 4 基团的形式存在。相比于硅氧间只能形成二氧连接, 硅碳间最多的四碳连接形式则可形成更为坚固的刚性骨架, 从而提高SiCO的模量、硬度、粘度、密度和玻璃转化温度。当体系中的自由C含量逐渐升高时, SiCO的弹性模量、弯曲强度、维氏硬度都会出现明显增加[63 -65 ] 。表2 比较了SiCO和玻璃态SiO2 之间的物理特性[64 ] 。在化学稳定性方面, SiCO经历如碱性条件或HF的腐蚀后, 由于其中的Si-C键可经受更高的亲核攻击(nucleophilic attack), 具有更高的无序和交联度, 并且与Si原子相连的C和自由C均可对腐蚀反应起阻碍作用, 因而比SiO2 表现出更好的化学稳定性[60 ] 。针对多孔SiCO的传热问题, Qiu等[66 ] 建立了一种描述其热传递的立方阵列交叉纳米球模型(cubic array of an intersecting nano-spheres model), 并发展了预测有效热导率的理论公式。结果表明, 多孔SiCO室温下的有效热导率在0.041~0.078 W/(m·K)之间, 该数值主要与SiCO固相体积分数以及相邻颗粒间接触长度(a )与颗粒直径(d )的比值a /d 有关。图5 给出了该研究中建立的多孔SiCO陶瓷的几何结构和热传递分析模型[66 ] 。
图5 多孔SiCO陶瓷的几何结构和热传递分析模型[66 ]
Fig. 5 Geometric structure and heat transfer analysis of macro-porous SiCO ceramics[66 ] (a) Cubic array of intersecting spherical structure; (b) Heat transfer in two contact spherical particles; (c) Heat transfer in sphere-gas-sphere structure
4 结束语
基于溶胶-凝胶法, 将具有多种存在形式的碳材料与SiO2 进行复合, 可形成具有不同气凝胶结构特征的C/SiO2 复合材料, 随后经高温碳热反应可转变为C/SiC复合材料。这种具有气凝胶结构特征的复合材料呈现出新的特性: C/SiO2 复合材料具有轻质、热稳定性高、吸附率高、抗高温辐射、高电催化活性等特性; C/SiC复合材料在继承C/SiO2 复合材料上述特性的同时, 还具有SiC材料自身的高硬度、高抗热震能力、高热稳定性等特点。碳材料与SiO2 之间的相对存在形式, 是决定该类复合材料性能的关键, 因而应该进一步关注: (1)改变碳源与硅源的引入方式, 借助两者各自呈现的特殊结构特征, 丰富其复合形式, 从而拓展复合材料的应用范围; (2)深入探讨制备过程中的反应机理, 研究碳/硅间的相互作用对复合材料形成的影响机制; (3)拓展气凝胶型多孔材料, 尤其是耐高温多孔材料的构成方法, 为其他类型材料的研究提供一种好的研究思路。
综上所述, 借助于硅与碳之间丰富的复合形式使具有气凝胶结构特征的C/SiO2 和C/SiC复合材料呈现出新的结构特征与特性, 为相关研究开辟了新的方向, 因而在高温隔热、吸附、催化、储氢、光电等多种领域具有广泛的应用前景和研究价值。
The authors have declared that no competing interests exist.
作者声明没有竞争性利益关系.
参考文献
文献选项
[1]
WHITE R J BRUN N BUDARIN V L et al. Always look on the “light” side of life: sustainable carbon aerogels.
ChemSusChem , 2014 , 7 (3 ): 670 -689 .
[本文引用: 2]
[2]
HUSING N SCHUBERT U. Aerogels-airy materials: chemistry, structure, and properties.
Angewandte Chemie International Edition , 1998 , 37 (1/2 ): 22 -45 .
[3]
PIERRE A C PAJONK G M. Chemistry of aerogels and their applications.
Chemical Reviews , 2002 , 102 (11 ): 4243 -4266 .
[4]
RAO A V BHAGAT S D HIRASHIMA H et al. Synthesis of flexible silica aerogels using methyltrimethoxysilane (MTMS) precursor.
Journal of Colloid and Interface Science , 2006 , 300 (1 ): 279 -285 .
[5]
RANDALL J P Tailoring mechanical properties of aerogels for aerospace applications.
ACS Applied Materials & Interfaces , 2011 , 3 (3 ): 613 -626 .
[6]
ANTONIETTI M Carbon aerogels and monoliths: control of porosity and nanoarchitecture via Sol-Gel routes.
Chemistry of Materials , 2013 , 26 (1 ): 196 -210 .
[7]
MALEKI H DURAES L PORTUGAL A. An overview on silica aerogels synthesis and different mechanical reinforcing strategies.
Journal of Non-Crystalline Solids , 2014 , 385 (2 ): 55 -74 .
[8]
WANG Z WANG D QIAN Z et al. Robust superhydrophobic bridged silsesquioxane aerogels with tunable performances and their applications.
ACS Applied Materials & Interfaces , 2015 , 7 (3 ): 2016 -2024 .
[本文引用: 1]
[9]
LEE D STEVENS P C ZENG S Q et al. Thermal characterization of carbon-opacified silica aerogels.
Journal of Non-Crystalline Solids , 1995 , 186 (2 ): 285 -290 .
[本文引用: 1]
[10]
ZHOU X F CUI S LIU Y et al. Adsorption capacity of hydrophobic SiO2 aerogel/activated carbon composite materials for TNT.
Science China Technological Sciences , 2013 , 56 (7 ): 1767 -1772 .
[本文引用: 2]
[11]
KONG Y ZHONG Y SHEN X et al. Synthesis and characterization of monolithic carbon/silicon carbide composite aerogels.
Journal of Porous Materials , 2013 , 20 (4 ): 845 -849 .
[本文引用: 2]
[12]
KONG Y SHEN X CUI S et al. Preparation of monolith SiC aerogel with high surface area and large pore volume and the structural evolution during the preparation.
Ceramics International , 2014 , 40 (6 ): 8265 -8271 .
[本文引用: 9]
[13]
HASEGAWA G KANAMORI K NAKANISHI K et al. A new route to monolithic macroporous SiC/C composites from biphenylene-bridged polysilsesquioxane gels.
Chemistry of Materials , 2010 , 22 (8 ): 2541 -2547 .
[本文引用: 2]
[14]
ERMAKOVA M A ERMAKOV D Y KUVSHINOV G G et al. Synthesis of high surface area silica gels using porous carbon matrices.
Journal of Porous Materials , 2000 , 7 (4 ): 435 -441 .
[本文引用: 1]
[15]
YE L JI Z H HAN W J et al. Synthesis and characterization of silica/carbon composite aerogels.
Journal of the American Ceramic Society , 2010 , 93 (4 ): 1156 -1163 .
[本文引用: 4]
[16]
SCHAEFER D W PEKALA R BEAUCAGE G. Origin of porosity in resorcinol-formaldehyde aerogels.
Journal of Non-Crystalline Solids , 1995 , 186 (2 ): 159 -167 .
[本文引用: 1]
[17]
TAMON H KITAMURA T OKAZAKI M. Preparation of silica aerogel from TEOS.
Journal of Colloid and Interface Science , 1998 , 197 (2 ): 353 -359 .
[18]
DU A ZHOU B ZHANG Z et al. A special material or a new state of matter: a review and reconsideration of the aerogel.
Materials , 2013 , 6 (3 ): 941 -968 .
[本文引用: 1]
[19]
AGUADO-SERRANO J Silica/C composites prepared by the Sol-Gel method. Influence of the synthesis parameters on textural characteristics.
Microporous and Mesoporous Materials , 2004 , 74 (74 ): 111 -119 .
[本文引用: 1]
[20]
LI X CHEN X SONG H. Synthesis of β-SiC nanostructures via the carbothermal reduction of resorcinol-formaldehyde/SiO2 hybrid aerogels.
Journal of Materials Science , 2009 , 44 (17 ): 4661 -4667 .
[本文引用: 2]
[21]
XU H ZHANG H HUANG Y et al. Porous carbon/silica composite monoliths derived from resorcinol-formaldehyde/TEOS.
Journal of Non-Crystalline Solids , 2010 , 356 (20/21/22 ): 971 -976 .
[本文引用: 1]
[22]
CHEN K BAO Z DU A et al. One-pot synthesis, characterization and properties of acid-catalyzed resorcinol/formaldehyde cross- linked silica aerogels and their conversion to hierarchical porous carbon monoliths.
Journal of Sol-Gel Science and Technology , 2012 , 62 (3 ): 294 -303 .
[本文引用: 4]
[23]
CHEN K BAO Z DU A et al. Synthesis of resorcinol-formaldehyde/ silica composite aerogels and their low-temperature conversion to mesoporous silicon carbide.
Microporous and Mesoporous Materials , 2012 , 149 (1 ): 16 -24 .
[本文引用: 2]
[24]
KONG Y ZHONG Y SHEN X et al. Facile synthesis of resorcinol-formaldehyde/silica composite aerogels and their transformation to monolithic carbon/silica and carbon/silicon carbide composite aerogels.
Journal of Non-Crystalline Solids , 2012 , 358 (23 ): 3150 -3155 .
[本文引用: 3]
[25]
KONG Y ZHONG Y SHEN X et al. Synthesis of monolithic mesoporous silicon carbide from resorcinol-formaldehyde/silica composites.
Materials Letters , 2013 , 99 (20 ): 108 -110 .
[本文引用: 2]
[26]
KONG Y ZHONG Y SHEN X et al. Effect of silica sources on nanostructures of resorcinol-formaldehyde/silica and carbon/silicon carbide composite aerogels.
Microporous and Mesoporous Materials , 2014 , 197 (10 ): 77 -82 .
[本文引用: 3]
[27]
ZHMUD B V SONNEFELD J. Aminopolysiloxane gels: production and properties.
Journal of Non-crystalline Solids , 1996 , 195 (1/2 ): 16 -27 .
[本文引用: 2]
[28]
YAO J WANG H ZHANG X et al. Role of pores in the carbothermal reduction of carbon-silica nanocomposites into silicon carbide nanostructures.
The Journal of Physical Chemistry C , 2007 , 111 (2 ): 636 -641 .
[本文引用: 5]
[29]
KIM H J KIM J H KIM W I et al. Nanoporous phloroglucinol- formaldehyde carbon aerogels for electrochemical use.
Korean Journal of Chemical Engineering , 2005 , 22 (22 ): 740 -744 .
[本文引用: 1]
[30]
SONG L FENG D LEE H J et al. Stabilizing surfactant templated cylindrical mesopores in polymer and carbon films through composite formation with silica reinforcement.
The Journal of Physical Chemistry C , 2010 , 114 (21 ): 9618 -9626 .
[本文引用: 1]
[31]
MEECHOONUCK M VAS-UMNUAY P PAVARAJAM V. Synthesis of porous silicon nitride using silica/carbon composite derived from phenol-resorcinol-formaldehyde gel.
Ceramics International , 2016 , 42 (9 ): 10879 -10885 .
[本文引用: 1]
[32]
ZHENG Y ZHENG Y LI Z et al. Preparations of C/SiC composites and their use as supports for Ru catalyst in ammonia synthesis.
Journal of Molecular Catalysis A: Chemical , 2009 , 301 (1/2 ): 79 -83 .
[本文引用: 1]
[33]
RAMAN V BAHL O P DHAWAN U. Synthesis of silicon carbide through the sol-gel process from different precursors.
Journal of Materials Science , 1995 , 30 (10 ): 2686 -2693 .
[本文引用: 1]
[34]
LI X K LIU L ZHANG Y X et al. Synthesis of nanometre silicon carbide whiskers from binary carbonaceous silica aerogels.
Carbon , 2001 , 39 (2 ): 159 -165 .
[本文引用: 1]
[35]
PREISS H BERGER L M BRAUN M. Formation of black glasses and silicon carbide from binary carbonaceous/silica hydrogels.
Carbon , 1995 , 33 (33 ): 1739 -1746 .
[本文引用: 1]
[36]
SERAJI M M GHAFOORIAN N S BAHRAMIAN A R et al. Preparation and characterization of C/SiO2 /SiC aerogels based on novolac/silica hybrid hyperporous materials.
Journal of Non- Crystalline Solids , 2015 , 425 (1 ): 146 -152 .
[本文引用: 1]
[37]
KARNIB M KABBANI A HOALIL H et al. Heavy metals removal using activated carbon, silica and silica activated carbon composite.
Energy Procedia , 2014 , 50 (1 ): 113 -120 .
[本文引用: 1]
[38]
LU X WANG P ARDUINI-SCHUSTER M C, et al. Thermal transport in organic and opacified silica monolithic aerogels.
Journal of Non-crystalline Solids , 1992 , 145 (1 ): 207 -210 .
[本文引用: 1]
[39]
LIU H LI T SHI Y et al. Thermal insulation composite prepared from carbon foam and silica aerogel under ambient pressure.
Journal of Materials Engineering and Performance , 2015 , 24 (10 ): 4054 -4059 .
[本文引用: 1]
[40]
PINCHUK O A DUNDAR F ATA A et al. Improved thermal stability, properties, and electrocatalytic activity of Sol-Gel silica modified carbon supported Pt catalysts.
International Journal of Hydrogen Energy , 2012 , 37 (3 ): 2111 -2120 .
[本文引用: 1]
[41]
MONER-GIRONA M MARTINEZ E ESTEVE J et al. Micromechanical properties of carbon-silica aerogel composites.
Applied Physics A , 2002 , 74 (1 ): 119 -122 .
[本文引用: 1]
[42]
LIU C KOMARNENI S. Carbon-silica xerogel and aerogel composites.
Journal of Porous Materials , 1995 , 1 (1 ): 75 -84 .
[本文引用: 3]
[43]
SPASSOVA I STOEVA N NICKOLOV R et al. Impact of carbon on the surface and activity of silica-carbon supported copper catalysts for reduction of nitrogen oxides.
Applied Surface Science , 2016 , 369 (1 ): 120 -129 .
[本文引用: 2]
[44]
WORSLEY M A KUNTZ J D SATCHER J H et al. Synthesis and characterization of monolithic, high surface area SiO2 /C and SiC/C composites.
Journal of Materials Chemistry , 2010 , 20 (23 ): 4840 -4844 .
[本文引用: 2]
[45]
LEVENTIS N SADEKAR A CHANDRASEKARANn N et al. Click synthesis of monolithic silicon carbide aerogels from polyacrylonitrile-coated 3D silica networks.
Chemistry of Materials , 2010 , 22 (9 ): 2790 -2803 .
[本文引用: 4]
[46]
PANTONO C G SINGH A K ZHANGH. Silicon oxycarbide glasses.
Journal of Sol-Gel Science and Technology , 1999 , 14 (1 ): 7 -25 .
[本文引用: 4]
[47]
LIU C CHEN H Z KOMARNENF S et al. High surface area SiC/silicon oxycarbide glasses prepared from phenyltrimethoxysilane- tetramethoxysilane gels.
Journal of Porous Materials , 1996 , 2 (3 ): 245 -252 .
[本文引用: 1]
[48]
SINGH A K PANTANO C G. Porous silicon oxycarbide glasses.
Journal of the American Ceramic Society , 1996 , 79 (10 ): 2696 -2704 .
[本文引用: 2]
[49]
ZHANG H PANTANO C G. Synthesis and characterization of silicon oxycarbide glasses.
Journal of the American Ceramic Society , 1990 , 73 (4 ): 958 -963 .
[本文引用: 1]
[50]
BABONNEAU F THORNE K MACKENZIE J D. Dimethyldiethoxysilane/tetraethoxysilane copolymers: precursors for the silicon-carbon-oxygen system.
Chemistry of Materials , 1989 , 1 (5 ): 554 -558 .
[本文引用: 3]
[51]
FENG J XIAO Y JIANG Y et al. Synthesis, structure, and properties of silicon oxycarbide aerogels derived from tetraethylortosilicate/polydimethylsiloxane.
Ceramics International , 2015 , 41 (4 ): 5281 -5286 .
[本文引用: 2]
[52]
TOURY B BLUM R GOLETTO V et al. Thermal stability of periodic mesoporous SiCO glasses.
Journal of Sol-Gel Science and Technology , 2005 , 33 (1 ): 99 -102 .
[本文引用: 3]
[53]
TAMAYO A TELLEZ L PENA-ALONSO R et al. Surface changes during pyrolytic conversion of hybrid materials to oxycarbide glasses.
Journal of Materials Science , 2009 , 44 (1 ): 5743 -5753 .
[本文引用: 3]
[54]
ARAVIND P R RATKE L KOLBE M et al. Gels dried under supercritical and ambient conditions: a comparative study and their subsequent conversion to silica-carbon composite aerogels.
Journal of Sol-Gel Science and Technology , 2013 , 67 (3 ): 592 -600 .
[本文引用: 1]
[55]
PRADEEP V S AYANA D G GRACZYK-ZAJAC M et al. High rate capability of SiOC ceramic aerogels with tailored porosity as anode materials for Li-ion batteries.
Electrochimica Acta , 2015 , 157 (1 ): 41 -45 .
[本文引用: 1]
[56]
ARAVIND P R SORARU G D. Porous silicon oxycarbide glasses from hybrid ambigels.
Microporous and Mesoporous Materials , 2011 , 142 (2/3 ): 511 -517 .
[本文引用: 1]
[57]
TAMAYO A RUBIO F RUBIO J et al. Surface and structural modification of nanostructured mesoporous silicon oxycarbide glasses obtained from preceramic hybrids aged in NH4 OH.
Journal of the American Ceramic Society , 2013 , 96 (1 ): 323 -330 .
[本文引用: 1]
[58]
PARMENTIER J SORARU G D BABONNEAU F. Influence of the microstructure on the high temperature behaviour of gel-derived SiOC glasses.
Journal of the European Ceramic Society , 2001 , 21 (6 ): 817 -824 .
[本文引用: 3]
[59]
WEINBERGER M PUCHEGGER S FROSCHL T et al. Sol- Gel Processing of a glycolated cyclic organosilane and its pyrolysis to silicon oxycarbide monoliths with multiscale porosity and large surface areas.
Chemistry of Materials , 2010 , 22 (4 ): 1509 -1520 .
[本文引用: 1]
[60]
SORARU G D MODENA S GUADAGNINO E et al. Chemical durability of silicon oxycarbide glasses.
Journal of the American Ceramic Society , 2002 , 85 (6 ): 1529 -1536 .
[本文引用: 3]
[61]
BREQUEL H PARMENTIER J WALTER S et al. Systematic structural characterization of the high-temperature behavior of nearly stoichiometric silicon oxycarbide glasses.
Chemistry of Materials , 2004 , 16 (1 ): 2585 -2598 .
[本文引用: 3]
[62]
LATOURNERIE J DEMPSEY P HOURLIER‐BAHLOUL D, et al. Silicon oxycarbide glasses: Part 1-Thermochemical stability.
Journal of the American Ceramic Society , 2006 , 89 (5 ): 1485 -1491 .
[本文引用: 1]
[63]
SORARU G D DALLAPICCOLA E D'ANDREA G. Mechanical characterization of Sol-Gel-derived silicon oxycarbide glasses.
Journal of the American Ceramic Society , 1996 , 79 (8 ): 2074 -2080 .
[本文引用: 1]
[64]
RENLUND G M PROCHAZKA S DOREMUS R H. Silicon oxycarbide glasses: Part II. Structure and properties.
Journal of Materials Research , 1991 , 6 (6 ): 2723 -2734 .
[本文引用: 1]
[65]
MOYSAN C RIEDEL R HARSHE R et al. Mechanical characterization of a polysiloxane-derived SiOC glass.
Journal of the European Ceramic Society , 2007 , 27 (1 ): 397 -403 .
[本文引用: 1]
[66]
QIU L LI Y M ZHENG X H et al. Thermal-conductivity studies of macro-porous polymer-derived SiOC ceramics.
International Journal of Thermophysics , 2014 , 35 (1 ): 76 -89 .
[本文引用: 4]
Always look on the “light” side of life: sustainable carbon aerogels.
2
2014
... 气凝胶是一类由纳米颗粒构成并呈现三维网络骨架结构的多孔固体材料[1 ] .由于构成骨架的固体颗粒和孔隙结构均为纳米量级, 因而这一独特结构使气凝胶呈现出如低密度、高孔隙率、高比表面积、低热导率、高透过率和高吸附率等特征和性能, 并可作为光学传感器、隔热材料、隔声材料、催化剂及载体、吸附剂、电极材料和惯性约束聚变靶材等使用, 具有广泛的应用前景[1 -8 ] . ...
... [1 -8 ]. ...
Aerogels-airy materials: chemistry, structure, and properties.
1998
Chemistry of aerogels and their applications.
2002
Synthesis of flexible silica aerogels using methyltrimethoxysilane (MTMS) precursor.
2006
Tailoring mechanical properties of aerogels for aerospace applications.
2011
Carbon aerogels and monoliths: control of porosity and nanoarchitecture via Sol-Gel routes.
2013
An overview on silica aerogels synthesis and different mechanical reinforcing strategies.
2014
Robust superhydrophobic bridged silsesquioxane aerogels with tunable performances and their applications.
1
2015
... 气凝胶是一类由纳米颗粒构成并呈现三维网络骨架结构的多孔固体材料[1 ] .由于构成骨架的固体颗粒和孔隙结构均为纳米量级, 因而这一独特结构使气凝胶呈现出如低密度、高孔隙率、高比表面积、低热导率、高透过率和高吸附率等特征和性能, 并可作为光学传感器、隔热材料、隔声材料、催化剂及载体、吸附剂、电极材料和惯性约束聚变靶材等使用, 具有广泛的应用前景[1 -8 ] . ...
Thermal characterization of carbon-opacified silica aerogels.
1
1995
... 除了单组份气凝胶材料以外, 双元或多元气凝胶则是结合不同组分间的优点形成具有新特性的复合气凝胶材料.其中, C/SiO2 复合气凝胶材料是研究较多并具有丰富复合结构形式的一类材料.根据应用背景的不同, 该复合材料可在保持某一组元特性的同时, 通过引入另一组元提高单一组元材料的性能.比如, 在SiO2 气凝胶中引入碳源作为红外遮光剂, 可以提高SiO2 气凝胶的抗辐射能力[9 ] ; 在活性炭中引入SiO2 气凝胶可以提高材料的吸附率[10 ] 等等.若对C/SiO2 复合气凝胶材料进行高温烧结, 经碳热还原(carbothermal reduction)反应, 可转化为C/SiC复合材料[11 ] 或SiC单体材料[12 ] .基于此方法获得的具有气凝胶结构特征的SiC材料, 除保持SiC本身的高硬度、高抗热震能力、高热导率、高热稳定性等特点外, 还具有高孔隙率、高比表面积等多孔材料的特征, 因而可在结构陶瓷、催化剂载体和过滤器等领域得以应用[12 -13 ] . ...
Adsorption capacity of hydrophobic SiO2 aerogel/activated carbon composite materials for TNT.
2
2013
... 除了单组份气凝胶材料以外, 双元或多元气凝胶则是结合不同组分间的优点形成具有新特性的复合气凝胶材料.其中, C/SiO2 复合气凝胶材料是研究较多并具有丰富复合结构形式的一类材料.根据应用背景的不同, 该复合材料可在保持某一组元特性的同时, 通过引入另一组元提高单一组元材料的性能.比如, 在SiO2 气凝胶中引入碳源作为红外遮光剂, 可以提高SiO2 气凝胶的抗辐射能力[9 ] ; 在活性炭中引入SiO2 气凝胶可以提高材料的吸附率[10 ] 等等.若对C/SiO2 复合气凝胶材料进行高温烧结, 经碳热还原(carbothermal reduction)反应, 可转化为C/SiC复合材料[11 ] 或SiC单体材料[12 ] .基于此方法获得的具有气凝胶结构特征的SiC材料, 除保持SiC本身的高硬度、高抗热震能力、高热导率、高热稳定性等特点外, 还具有高孔隙率、高比表面积等多孔材料的特征, 因而可在结构陶瓷、催化剂载体和过滤器等领域得以应用[12 -13 ] . ...
... 在吸附领域, 活性炭经SiO2 溶胶改性后, 借助于SiO2 气凝胶纳米多孔的结构特性, 可以提高活性炭材料的比表面积和孔隙率, 进而提高活性炭的吸附率.Zhou等[10 ] 将活性炭浸入以TEOS和苯基三乙氧基硅烷(phenyltriethoxysilane, PTES)或甲基三乙氧基硅烷(methyltriethoxysilane, MTES)为混合先驱体制备的SiO2 溶胶中, SiO2 溶胶颗粒原位聚合于碳材料表面, 获得了SiO2 /活性炭复合材料.该材料的微观结构上是SiO2 气凝胶包裹活性炭颗粒, 且骨架结构比较松散.依靠表面吸附(surface adsorption)作用, 将其吸附率从活性炭的76%提高到96.5%.该材料可实现从废水中吸附三硝基甲苯(2,4,6-trinitrotoluene, TNT)的作用.同时, 有机硅氧烷先驱体提供的不同疏水基团对吸附率也有影响.Karnib等[37 ] 用APTES对SiO2 颗粒进行表面修饰后, 与活性炭在溶液状态下混合, 经干燥和研磨后, 获得的SiO2 /活性炭复合颗粒对水溶液中的重金属离子具有很好的吸附效果. ...
Synthesis and characterization of monolithic carbon/silicon carbide composite aerogels.
2
2013
... 除了单组份气凝胶材料以外, 双元或多元气凝胶则是结合不同组分间的优点形成具有新特性的复合气凝胶材料.其中, C/SiO2 复合气凝胶材料是研究较多并具有丰富复合结构形式的一类材料.根据应用背景的不同, 该复合材料可在保持某一组元特性的同时, 通过引入另一组元提高单一组元材料的性能.比如, 在SiO2 气凝胶中引入碳源作为红外遮光剂, 可以提高SiO2 气凝胶的抗辐射能力[9 ] ; 在活性炭中引入SiO2 气凝胶可以提高材料的吸附率[10 ] 等等.若对C/SiO2 复合气凝胶材料进行高温烧结, 经碳热还原(carbothermal reduction)反应, 可转化为C/SiC复合材料[11 ] 或SiC单体材料[12 ] .基于此方法获得的具有气凝胶结构特征的SiC材料, 除保持SiC本身的高硬度、高抗热震能力、高热导率、高热稳定性等特点外, 还具有高孔隙率、高比表面积等多孔材料的特征, 因而可在结构陶瓷、催化剂载体和过滤器等领域得以应用[12 -13 ] . ...
... 基于上述考虑, 当采用正硅酸乙酯(tetraethoxysilane, TEOS)为先驱体水解得到SiO2 溶胶, 加入到事先配制的RF溶胶中, 可以获得均匀的混合溶胶.混合溶胶经凝胶、干燥后获得RF/SiO2 气凝胶, 随后在惰性气体保护下热解得到C/SiO2 气凝胶材料[11 -12 ,19 -20 ] .TEOS加入RF溶胶能明显缩短体系的凝胶时间, 但是随着TEOS含量的逐渐增加, 气凝胶孔径逐渐减小, 孔内毛细张力增大, 气凝胶易于开裂[21 ] .Chen等[22 -23 ] 针对该混合溶胶体系, 采用一步酸催化快速合成法(one-pot acid-catalyzed rapid synthesis route)制备了RF/SiO2 气凝胶.在该方法中, HCl取代Na2 CO3 作为酸性催化剂, 可以提高芳香族链亲电取代(electrophilic aromatic substitution)的活性, 从而加速芳香族链之间亚甲基桥联的形成; 乙腈(CH3 CN)取代乙醇作为溶剂, 为TEOS的溶胶-凝胶过程提供了更加合适的环境, 适合于一步形成RF/ SiO2 体系.这种在CH3 CN溶剂中采用酸性催化剂的反应体系可以显著加快RF先驱体的反应速率, 可以将混合体系的凝胶时间由几天缩短至几个小时.RF/SiO2 复合气凝胶材料的孔结构呈现与纯SiO2 和纯RF气凝胶相似的结构特征, 并具有低的热导率和高的力学行为.图2 给出了酸催化RF/SiO2 凝胶的形成过程[22 ] . ...
Preparation of monolith SiC aerogel with high surface area and large pore volume and the structural evolution during the preparation.
9
2014
... 除了单组份气凝胶材料以外, 双元或多元气凝胶则是结合不同组分间的优点形成具有新特性的复合气凝胶材料.其中, C/SiO2 复合气凝胶材料是研究较多并具有丰富复合结构形式的一类材料.根据应用背景的不同, 该复合材料可在保持某一组元特性的同时, 通过引入另一组元提高单一组元材料的性能.比如, 在SiO2 气凝胶中引入碳源作为红外遮光剂, 可以提高SiO2 气凝胶的抗辐射能力[9 ] ; 在活性炭中引入SiO2 气凝胶可以提高材料的吸附率[10 ] 等等.若对C/SiO2 复合气凝胶材料进行高温烧结, 经碳热还原(carbothermal reduction)反应, 可转化为C/SiC复合材料[11 ] 或SiC单体材料[12 ] .基于此方法获得的具有气凝胶结构特征的SiC材料, 除保持SiC本身的高硬度、高抗热震能力、高热导率、高热稳定性等特点外, 还具有高孔隙率、高比表面积等多孔材料的特征, 因而可在结构陶瓷、催化剂载体和过滤器等领域得以应用[12 -13 ] . ...
... [12 -13 ]. ...
... 共聚法是指硅源和碳源分别以SiO2 溶胶和有机溶胶的形式引入, 经均匀混合、凝胶和干燥后, 形成有机-SiO2 气凝胶, 再经高温烧结获得C/SiO2 和C/SiC复合材料.该方法通常采用有机硅氧烷醇盐为先驱体配制SiO2 溶胶, 而碳源通常以间苯二酚(resorcinol, R)和甲醛(formaldehyde, F)聚合物先驱体的形式引入, 经水解和缩聚后形成间苯二酚-甲醛(resorcinol-formaldehyde, RF)溶胶.凝胶后, 形成的混合凝胶中有机骨架与SiO2 骨架相互交叉缠绕, 但通常这两种网络骨架之间并无化学键连接.由于RF气凝胶的溶胶-凝胶过程相较于SiO2 十分缓慢[16 -18 ] , 因而RF与SiO2 凝胶形成时间的不同往往会在SiO2 凝胶骨架表面形成RF凝胶.为了更好地获得均匀的RF-SiO2 气凝胶, 通常采用分别溶胶再混合凝胶的方法制备RF-SiO2 气凝胶.图1 给出了RF-SiO2 气凝胶的形成过程及其向SiC气凝胶的转变示意图[12 ] . ...
... RF-SiO2 气凝胶的形成过程及其向SiC气凝胶的转变示意图[12 ] ...
... Formation of RF-SiO2 aerogel and its transformation to SiC aerogel[12 ] ...
... 基于上述考虑, 当采用正硅酸乙酯(tetraethoxysilane, TEOS)为先驱体水解得到SiO2 溶胶, 加入到事先配制的RF溶胶中, 可以获得均匀的混合溶胶.混合溶胶经凝胶、干燥后获得RF/SiO2 气凝胶, 随后在惰性气体保护下热解得到C/SiO2 气凝胶材料[11 -12 ,19 -20 ] .TEOS加入RF溶胶能明显缩短体系的凝胶时间, 但是随着TEOS含量的逐渐增加, 气凝胶孔径逐渐减小, 孔内毛细张力增大, 气凝胶易于开裂[21 ] .Chen等[22 -23 ] 针对该混合溶胶体系, 采用一步酸催化快速合成法(one-pot acid-catalyzed rapid synthesis route)制备了RF/SiO2 气凝胶.在该方法中, HCl取代Na2 CO3 作为酸性催化剂, 可以提高芳香族链亲电取代(electrophilic aromatic substitution)的活性, 从而加速芳香族链之间亚甲基桥联的形成; 乙腈(CH3 CN)取代乙醇作为溶剂, 为TEOS的溶胶-凝胶过程提供了更加合适的环境, 适合于一步形成RF/ SiO2 体系.这种在CH3 CN溶剂中采用酸性催化剂的反应体系可以显著加快RF先驱体的反应速率, 可以将混合体系的凝胶时间由几天缩短至几个小时.RF/SiO2 复合气凝胶材料的孔结构呈现与纯SiO2 和纯RF气凝胶相似的结构特征, 并具有低的热导率和高的力学行为.图2 给出了酸催化RF/SiO2 凝胶的形成过程[22 ] . ...
... 当热解温度提高至1200~1500℃, RF/SiO2 气凝胶或C/SiO2 气凝胶经碳热还原反应转变为SiC或SiC/C气凝胶, 总的反应方程如式(1)所示[12 ,20 ] : ...
... 在这一过程中, SiC首先在先驱体表面形核(nuclei), 随后长大成纳米晶须(nanowhiskers).其间相互贯穿孔的存在为气体产物(如SiO、CO和CO2 )的扩散提供了通路, 加速了这一反应的进行[12 ] .C与SiO2 的比例和C/SiO2 复合材料的致密度是影响SiC反应动力学和最终SiC微观结构的重要因素.图3 给出了不同结构SiC的形成过程示意图[28 ] .高碳硅比例下, 介孔C/SiO2 可形成纳米级纤维状和颗粒状SiC, 如图3 (a)所示; 低碳硅比例下, 除形成纳米纤维状和颗粒状SiC以外, 还渗透着少量自由C, 如图3 (b)所示.其中, 纤维状和颗粒状SiC产物的比例可通过改变碳硅间的比例并利用C的渗透加以调节.高碳硅比例的致密C/SiO2 可转变为介孔SiC, 如图3 (c)所示.这是由于高碳硅比例以及C/SiO2 和C之间界面结构可以提高碳热还原反应速率, 介孔在为气态产物提供扩散通道的同时, 也会限制介孔C/SiO2 材料中SiC的生长[28 ] .除此以外, 采用低温镁热反应(low-temperature magnesiothermic reaction)可使C/SiO2 材料在700℃的低温下转变成SiC[23 ] .通过碳热还原RF/SiO2 气凝胶制得的材料是以β- SiC为主体, 并含有少量α-SiC的SiC或C/SiC气凝胶材料.该材料具有多孔结构和高的比表面积, 并具有高硬度、好的抗热震能力、高热导和高稳定性、低膨胀系数和大的带隙, 可用于增强相、催化剂、高能和高频电子材料、光电材料、抗辐射材料、氢分离膜载体和吸波器件等[12 ] . ...
... [12 ]. ...
A new route to monolithic macroporous SiC/C composites from biphenylene-bridged polysilsesquioxane gels.
2
2010
... 除了单组份气凝胶材料以外, 双元或多元气凝胶则是结合不同组分间的优点形成具有新特性的复合气凝胶材料.其中, C/SiO2 复合气凝胶材料是研究较多并具有丰富复合结构形式的一类材料.根据应用背景的不同, 该复合材料可在保持某一组元特性的同时, 通过引入另一组元提高单一组元材料的性能.比如, 在SiO2 气凝胶中引入碳源作为红外遮光剂, 可以提高SiO2 气凝胶的抗辐射能力[9 ] ; 在活性炭中引入SiO2 气凝胶可以提高材料的吸附率[10 ] 等等.若对C/SiO2 复合气凝胶材料进行高温烧结, 经碳热还原(carbothermal reduction)反应, 可转化为C/SiC复合材料[11 ] 或SiC单体材料[12 ] .基于此方法获得的具有气凝胶结构特征的SiC材料, 除保持SiC本身的高硬度、高抗热震能力、高热导率、高热稳定性等特点外, 还具有高孔隙率、高比表面积等多孔材料的特征, 因而可在结构陶瓷、催化剂载体和过滤器等领域得以应用[12 -13 ] . ...
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
Synthesis of high surface area silica gels using porous carbon matrices.
1
2000
... 通常, 基于溶胶-凝胶法制备的C/SiO2 和C/SiC复合材料具有气凝胶材料的结构特征.碳化前, 借助于均匀分布的单元或双元溶胶胶体颗粒实现硅元素与碳元素的复合.一般而言, SiO2 常以有机硅氧烷为先驱体经水解缩聚后形成的SiO2 溶胶方式引入.而对于碳源的引入, 由于碳材料的多样性, 如催化丝状碳(catalytic filamentous carbon, CFC)、Sibunit碳、颗粒状碳黑(granular carbon black)、活性炭(activated carbon)、碳编织体(carbon weave)、碳毡(carbon felt)[14 ] 以及由有机聚合物提供的碳源[15 ] 等, 碳的引入呈现出复杂的形式.根据硅源与碳源引入方式的不同, 本文将基于溶胶-凝胶法制备的具有气凝胶材料结构特征的C/SiO2 和C/SiC复合材料的制备方式主要分为三类, 即共聚法(co-polymerization)、浸入法(impregnation)和聚合物先驱体热解法(polymeric precursors pyrolysis). ...
Synthesis and characterization of silica/carbon composite aerogels.
4
2010
... 通常, 基于溶胶-凝胶法制备的C/SiO2 和C/SiC复合材料具有气凝胶材料的结构特征.碳化前, 借助于均匀分布的单元或双元溶胶胶体颗粒实现硅元素与碳元素的复合.一般而言, SiO2 常以有机硅氧烷为先驱体经水解缩聚后形成的SiO2 溶胶方式引入.而对于碳源的引入, 由于碳材料的多样性, 如催化丝状碳(catalytic filamentous carbon, CFC)、Sibunit碳、颗粒状碳黑(granular carbon black)、活性炭(activated carbon)、碳编织体(carbon weave)、碳毡(carbon felt)[14 ] 以及由有机聚合物提供的碳源[15 ] 等, 碳的引入呈现出复杂的形式.根据硅源与碳源引入方式的不同, 本文将基于溶胶-凝胶法制备的具有气凝胶材料结构特征的C/SiO2 和C/SiC复合材料的制备方式主要分为三类, 即共聚法(co-polymerization)、浸入法(impregnation)和聚合物先驱体热解法(polymeric precursors pyrolysis). ...
... 为了避免以TEOS或正硅酸甲酯(tetramethy lorthosilicate, TMOS)为硅源制备RF/SiO2 气凝胶时的复杂溶胶-凝胶过程, 进一步减少水解缩聚的时间, 有研究采用硅烷偶联剂作为硅源一步合成RF/ SiO2 气凝胶[15 ,24 -26 ] .在该类方法中, 硅烷偶联剂3-氨基丙基三乙氧基硅烷[(3-aminopropyl)triethoxysilane, APTES]可作为硅源, 直接与R和F混合形成溶胶, 凝胶并超临界干燥后获得RF/SiO2 复合气凝胶[15 ,24 -25 ] .APTES含量的增加将减少混合溶胶体系的凝胶时间[24 ,27 ] .在该溶胶体系中, APTES除了作为硅源先驱体参与水解缩聚反应形成Si-O-Si网络结构以外, 还可作为“内部催化剂(internal catalyst)”促进R和F之间的缩聚反应.这是由于APTES中氨基的水合作用(hydration of amino groups)在APTES水解产物中形成了环状的分子间氢键, 为反应提供了良好的通路, 从而加速溶胶-凝胶过程[27 ] .RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
... [15 ,24 -25 ].APTES含量的增加将减少混合溶胶体系的凝胶时间[24 ,27 ] .在该溶胶体系中, APTES除了作为硅源先驱体参与水解缩聚反应形成Si-O-Si网络结构以外, 还可作为“内部催化剂(internal catalyst)”促进R和F之间的缩聚反应.这是由于APTES中氨基的水合作用(hydration of amino groups)在APTES水解产物中形成了环状的分子间氢键, 为反应提供了良好的通路, 从而加速溶胶-凝胶过程[27 ] .RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
... [15 ]), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
Origin of porosity in resorcinol-formaldehyde aerogels.
1
1995
... 共聚法是指硅源和碳源分别以SiO2 溶胶和有机溶胶的形式引入, 经均匀混合、凝胶和干燥后, 形成有机-SiO2 气凝胶, 再经高温烧结获得C/SiO2 和C/SiC复合材料.该方法通常采用有机硅氧烷醇盐为先驱体配制SiO2 溶胶, 而碳源通常以间苯二酚(resorcinol, R)和甲醛(formaldehyde, F)聚合物先驱体的形式引入, 经水解和缩聚后形成间苯二酚-甲醛(resorcinol-formaldehyde, RF)溶胶.凝胶后, 形成的混合凝胶中有机骨架与SiO2 骨架相互交叉缠绕, 但通常这两种网络骨架之间并无化学键连接.由于RF气凝胶的溶胶-凝胶过程相较于SiO2 十分缓慢[16 -18 ] , 因而RF与SiO2 凝胶形成时间的不同往往会在SiO2 凝胶骨架表面形成RF凝胶.为了更好地获得均匀的RF-SiO2 气凝胶, 通常采用分别溶胶再混合凝胶的方法制备RF-SiO2 气凝胶.图1 给出了RF-SiO2 气凝胶的形成过程及其向SiC气凝胶的转变示意图[12 ] . ...
Preparation of silica aerogel from TEOS.
1998
A special material or a new state of matter: a review and reconsideration of the aerogel.
1
2013
... 共聚法是指硅源和碳源分别以SiO2 溶胶和有机溶胶的形式引入, 经均匀混合、凝胶和干燥后, 形成有机-SiO2 气凝胶, 再经高温烧结获得C/SiO2 和C/SiC复合材料.该方法通常采用有机硅氧烷醇盐为先驱体配制SiO2 溶胶, 而碳源通常以间苯二酚(resorcinol, R)和甲醛(formaldehyde, F)聚合物先驱体的形式引入, 经水解和缩聚后形成间苯二酚-甲醛(resorcinol-formaldehyde, RF)溶胶.凝胶后, 形成的混合凝胶中有机骨架与SiO2 骨架相互交叉缠绕, 但通常这两种网络骨架之间并无化学键连接.由于RF气凝胶的溶胶-凝胶过程相较于SiO2 十分缓慢[16 -18 ] , 因而RF与SiO2 凝胶形成时间的不同往往会在SiO2 凝胶骨架表面形成RF凝胶.为了更好地获得均匀的RF-SiO2 气凝胶, 通常采用分别溶胶再混合凝胶的方法制备RF-SiO2 气凝胶.图1 给出了RF-SiO2 气凝胶的形成过程及其向SiC气凝胶的转变示意图[12 ] . ...
Silica/C composites prepared by the Sol-Gel method. Influence of the synthesis parameters on textural characteristics.
1
2004
... 基于上述考虑, 当采用正硅酸乙酯(tetraethoxysilane, TEOS)为先驱体水解得到SiO2 溶胶, 加入到事先配制的RF溶胶中, 可以获得均匀的混合溶胶.混合溶胶经凝胶、干燥后获得RF/SiO2 气凝胶, 随后在惰性气体保护下热解得到C/SiO2 气凝胶材料[11 -12 ,19 -20 ] .TEOS加入RF溶胶能明显缩短体系的凝胶时间, 但是随着TEOS含量的逐渐增加, 气凝胶孔径逐渐减小, 孔内毛细张力增大, 气凝胶易于开裂[21 ] .Chen等[22 -23 ] 针对该混合溶胶体系, 采用一步酸催化快速合成法(one-pot acid-catalyzed rapid synthesis route)制备了RF/SiO2 气凝胶.在该方法中, HCl取代Na2 CO3 作为酸性催化剂, 可以提高芳香族链亲电取代(electrophilic aromatic substitution)的活性, 从而加速芳香族链之间亚甲基桥联的形成; 乙腈(CH3 CN)取代乙醇作为溶剂, 为TEOS的溶胶-凝胶过程提供了更加合适的环境, 适合于一步形成RF/ SiO2 体系.这种在CH3 CN溶剂中采用酸性催化剂的反应体系可以显著加快RF先驱体的反应速率, 可以将混合体系的凝胶时间由几天缩短至几个小时.RF/SiO2 复合气凝胶材料的孔结构呈现与纯SiO2 和纯RF气凝胶相似的结构特征, 并具有低的热导率和高的力学行为.图2 给出了酸催化RF/SiO2 凝胶的形成过程[22 ] . ...
Synthesis of β-SiC nanostructures via the carbothermal reduction of resorcinol-formaldehyde/SiO2 hybrid aerogels.
2
2009
... 基于上述考虑, 当采用正硅酸乙酯(tetraethoxysilane, TEOS)为先驱体水解得到SiO2 溶胶, 加入到事先配制的RF溶胶中, 可以获得均匀的混合溶胶.混合溶胶经凝胶、干燥后获得RF/SiO2 气凝胶, 随后在惰性气体保护下热解得到C/SiO2 气凝胶材料[11 -12 ,19 -20 ] .TEOS加入RF溶胶能明显缩短体系的凝胶时间, 但是随着TEOS含量的逐渐增加, 气凝胶孔径逐渐减小, 孔内毛细张力增大, 气凝胶易于开裂[21 ] .Chen等[22 -23 ] 针对该混合溶胶体系, 采用一步酸催化快速合成法(one-pot acid-catalyzed rapid synthesis route)制备了RF/SiO2 气凝胶.在该方法中, HCl取代Na2 CO3 作为酸性催化剂, 可以提高芳香族链亲电取代(electrophilic aromatic substitution)的活性, 从而加速芳香族链之间亚甲基桥联的形成; 乙腈(CH3 CN)取代乙醇作为溶剂, 为TEOS的溶胶-凝胶过程提供了更加合适的环境, 适合于一步形成RF/ SiO2 体系.这种在CH3 CN溶剂中采用酸性催化剂的反应体系可以显著加快RF先驱体的反应速率, 可以将混合体系的凝胶时间由几天缩短至几个小时.RF/SiO2 复合气凝胶材料的孔结构呈现与纯SiO2 和纯RF气凝胶相似的结构特征, 并具有低的热导率和高的力学行为.图2 给出了酸催化RF/SiO2 凝胶的形成过程[22 ] . ...
... 当热解温度提高至1200~1500℃, RF/SiO2 气凝胶或C/SiO2 气凝胶经碳热还原反应转变为SiC或SiC/C气凝胶, 总的反应方程如式(1)所示[12 ,20 ] : ...
Porous carbon/silica composite monoliths derived from resorcinol-formaldehyde/TEOS.
1
2010
... 基于上述考虑, 当采用正硅酸乙酯(tetraethoxysilane, TEOS)为先驱体水解得到SiO2 溶胶, 加入到事先配制的RF溶胶中, 可以获得均匀的混合溶胶.混合溶胶经凝胶、干燥后获得RF/SiO2 气凝胶, 随后在惰性气体保护下热解得到C/SiO2 气凝胶材料[11 -12 ,19 -20 ] .TEOS加入RF溶胶能明显缩短体系的凝胶时间, 但是随着TEOS含量的逐渐增加, 气凝胶孔径逐渐减小, 孔内毛细张力增大, 气凝胶易于开裂[21 ] .Chen等[22 -23 ] 针对该混合溶胶体系, 采用一步酸催化快速合成法(one-pot acid-catalyzed rapid synthesis route)制备了RF/SiO2 气凝胶.在该方法中, HCl取代Na2 CO3 作为酸性催化剂, 可以提高芳香族链亲电取代(electrophilic aromatic substitution)的活性, 从而加速芳香族链之间亚甲基桥联的形成; 乙腈(CH3 CN)取代乙醇作为溶剂, 为TEOS的溶胶-凝胶过程提供了更加合适的环境, 适合于一步形成RF/ SiO2 体系.这种在CH3 CN溶剂中采用酸性催化剂的反应体系可以显著加快RF先驱体的反应速率, 可以将混合体系的凝胶时间由几天缩短至几个小时.RF/SiO2 复合气凝胶材料的孔结构呈现与纯SiO2 和纯RF气凝胶相似的结构特征, 并具有低的热导率和高的力学行为.图2 给出了酸催化RF/SiO2 凝胶的形成过程[22 ] . ...
One-pot synthesis, characterization and properties of acid-catalyzed resorcinol/formaldehyde cross- linked silica aerogels and their conversion to hierarchical porous carbon monoliths.
4
2012
... 基于上述考虑, 当采用正硅酸乙酯(tetraethoxysilane, TEOS)为先驱体水解得到SiO2 溶胶, 加入到事先配制的RF溶胶中, 可以获得均匀的混合溶胶.混合溶胶经凝胶、干燥后获得RF/SiO2 气凝胶, 随后在惰性气体保护下热解得到C/SiO2 气凝胶材料[11 -12 ,19 -20 ] .TEOS加入RF溶胶能明显缩短体系的凝胶时间, 但是随着TEOS含量的逐渐增加, 气凝胶孔径逐渐减小, 孔内毛细张力增大, 气凝胶易于开裂[21 ] .Chen等[22 -23 ] 针对该混合溶胶体系, 采用一步酸催化快速合成法(one-pot acid-catalyzed rapid synthesis route)制备了RF/SiO2 气凝胶.在该方法中, HCl取代Na2 CO3 作为酸性催化剂, 可以提高芳香族链亲电取代(electrophilic aromatic substitution)的活性, 从而加速芳香族链之间亚甲基桥联的形成; 乙腈(CH3 CN)取代乙醇作为溶剂, 为TEOS的溶胶-凝胶过程提供了更加合适的环境, 适合于一步形成RF/ SiO2 体系.这种在CH3 CN溶剂中采用酸性催化剂的反应体系可以显著加快RF先驱体的反应速率, 可以将混合体系的凝胶时间由几天缩短至几个小时.RF/SiO2 复合气凝胶材料的孔结构呈现与纯SiO2 和纯RF气凝胶相似的结构特征, 并具有低的热导率和高的力学行为.图2 给出了酸催化RF/SiO2 凝胶的形成过程[22 ] . ...
... [22 ]. ...
... 酸催化RF/SiO2 凝胶的形成过程示意图[22 ] ...
... Formation of acidcatalyzed RF/SiO2 hybrid gels[22 ] ...
Synthesis of resorcinol-formaldehyde/ silica composite aerogels and their low-temperature conversion to mesoporous silicon carbide.
2
2012
... 基于上述考虑, 当采用正硅酸乙酯(tetraethoxysilane, TEOS)为先驱体水解得到SiO2 溶胶, 加入到事先配制的RF溶胶中, 可以获得均匀的混合溶胶.混合溶胶经凝胶、干燥后获得RF/SiO2 气凝胶, 随后在惰性气体保护下热解得到C/SiO2 气凝胶材料[11 -12 ,19 -20 ] .TEOS加入RF溶胶能明显缩短体系的凝胶时间, 但是随着TEOS含量的逐渐增加, 气凝胶孔径逐渐减小, 孔内毛细张力增大, 气凝胶易于开裂[21 ] .Chen等[22 -23 ] 针对该混合溶胶体系, 采用一步酸催化快速合成法(one-pot acid-catalyzed rapid synthesis route)制备了RF/SiO2 气凝胶.在该方法中, HCl取代Na2 CO3 作为酸性催化剂, 可以提高芳香族链亲电取代(electrophilic aromatic substitution)的活性, 从而加速芳香族链之间亚甲基桥联的形成; 乙腈(CH3 CN)取代乙醇作为溶剂, 为TEOS的溶胶-凝胶过程提供了更加合适的环境, 适合于一步形成RF/ SiO2 体系.这种在CH3 CN溶剂中采用酸性催化剂的反应体系可以显著加快RF先驱体的反应速率, 可以将混合体系的凝胶时间由几天缩短至几个小时.RF/SiO2 复合气凝胶材料的孔结构呈现与纯SiO2 和纯RF气凝胶相似的结构特征, 并具有低的热导率和高的力学行为.图2 给出了酸催化RF/SiO2 凝胶的形成过程[22 ] . ...
... 在这一过程中, SiC首先在先驱体表面形核(nuclei), 随后长大成纳米晶须(nanowhiskers).其间相互贯穿孔的存在为气体产物(如SiO、CO和CO2 )的扩散提供了通路, 加速了这一反应的进行[12 ] .C与SiO2 的比例和C/SiO2 复合材料的致密度是影响SiC反应动力学和最终SiC微观结构的重要因素.图3 给出了不同结构SiC的形成过程示意图[28 ] .高碳硅比例下, 介孔C/SiO2 可形成纳米级纤维状和颗粒状SiC, 如图3 (a)所示; 低碳硅比例下, 除形成纳米纤维状和颗粒状SiC以外, 还渗透着少量自由C, 如图3 (b)所示.其中, 纤维状和颗粒状SiC产物的比例可通过改变碳硅间的比例并利用C的渗透加以调节.高碳硅比例的致密C/SiO2 可转变为介孔SiC, 如图3 (c)所示.这是由于高碳硅比例以及C/SiO2 和C之间界面结构可以提高碳热还原反应速率, 介孔在为气态产物提供扩散通道的同时, 也会限制介孔C/SiO2 材料中SiC的生长[28 ] .除此以外, 采用低温镁热反应(low-temperature magnesiothermic reaction)可使C/SiO2 材料在700℃的低温下转变成SiC[23 ] .通过碳热还原RF/SiO2 气凝胶制得的材料是以β- SiC为主体, 并含有少量α-SiC的SiC或C/SiC气凝胶材料.该材料具有多孔结构和高的比表面积, 并具有高硬度、好的抗热震能力、高热导和高稳定性、低膨胀系数和大的带隙, 可用于增强相、催化剂、高能和高频电子材料、光电材料、抗辐射材料、氢分离膜载体和吸波器件等[12 ] . ...
Facile synthesis of resorcinol-formaldehyde/silica composite aerogels and their transformation to monolithic carbon/silica and carbon/silicon carbide composite aerogels.
3
2012
... 为了避免以TEOS或正硅酸甲酯(tetramethy lorthosilicate, TMOS)为硅源制备RF/SiO2 气凝胶时的复杂溶胶-凝胶过程, 进一步减少水解缩聚的时间, 有研究采用硅烷偶联剂作为硅源一步合成RF/ SiO2 气凝胶[15 ,24 -26 ] .在该类方法中, 硅烷偶联剂3-氨基丙基三乙氧基硅烷[(3-aminopropyl)triethoxysilane, APTES]可作为硅源, 直接与R和F混合形成溶胶, 凝胶并超临界干燥后获得RF/SiO2 复合气凝胶[15 ,24 -25 ] .APTES含量的增加将减少混合溶胶体系的凝胶时间[24 ,27 ] .在该溶胶体系中, APTES除了作为硅源先驱体参与水解缩聚反应形成Si-O-Si网络结构以外, 还可作为“内部催化剂(internal catalyst)”促进R和F之间的缩聚反应.这是由于APTES中氨基的水合作用(hydration of amino groups)在APTES水解产物中形成了环状的分子间氢键, 为反应提供了良好的通路, 从而加速溶胶-凝胶过程[27 ] .RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
... ,24 -25 ].APTES含量的增加将减少混合溶胶体系的凝胶时间[24 ,27 ] .在该溶胶体系中, APTES除了作为硅源先驱体参与水解缩聚反应形成Si-O-Si网络结构以外, 还可作为“内部催化剂(internal catalyst)”促进R和F之间的缩聚反应.这是由于APTES中氨基的水合作用(hydration of amino groups)在APTES水解产物中形成了环状的分子间氢键, 为反应提供了良好的通路, 从而加速溶胶-凝胶过程[27 ] .RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
... [24 ,27 ].在该溶胶体系中, APTES除了作为硅源先驱体参与水解缩聚反应形成Si-O-Si网络结构以外, 还可作为“内部催化剂(internal catalyst)”促进R和F之间的缩聚反应.这是由于APTES中氨基的水合作用(hydration of amino groups)在APTES水解产物中形成了环状的分子间氢键, 为反应提供了良好的通路, 从而加速溶胶-凝胶过程[27 ] .RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
Synthesis of monolithic mesoporous silicon carbide from resorcinol-formaldehyde/silica composites.
2
2013
... 为了避免以TEOS或正硅酸甲酯(tetramethy lorthosilicate, TMOS)为硅源制备RF/SiO2 气凝胶时的复杂溶胶-凝胶过程, 进一步减少水解缩聚的时间, 有研究采用硅烷偶联剂作为硅源一步合成RF/ SiO2 气凝胶[15 ,24 -26 ] .在该类方法中, 硅烷偶联剂3-氨基丙基三乙氧基硅烷[(3-aminopropyl)triethoxysilane, APTES]可作为硅源, 直接与R和F混合形成溶胶, 凝胶并超临界干燥后获得RF/SiO2 复合气凝胶[15 ,24 -25 ] .APTES含量的增加将减少混合溶胶体系的凝胶时间[24 ,27 ] .在该溶胶体系中, APTES除了作为硅源先驱体参与水解缩聚反应形成Si-O-Si网络结构以外, 还可作为“内部催化剂(internal catalyst)”促进R和F之间的缩聚反应.这是由于APTES中氨基的水合作用(hydration of amino groups)在APTES水解产物中形成了环状的分子间氢键, 为反应提供了良好的通路, 从而加速溶胶-凝胶过程[27 ] .RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
... [25 ].以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
Effect of silica sources on nanostructures of resorcinol-formaldehyde/silica and carbon/silicon carbide composite aerogels.
3
2014
... 为了避免以TEOS或正硅酸甲酯(tetramethy lorthosilicate, TMOS)为硅源制备RF/SiO2 气凝胶时的复杂溶胶-凝胶过程, 进一步减少水解缩聚的时间, 有研究采用硅烷偶联剂作为硅源一步合成RF/ SiO2 气凝胶[15 ,24 -26 ] .在该类方法中, 硅烷偶联剂3-氨基丙基三乙氧基硅烷[(3-aminopropyl)triethoxysilane, APTES]可作为硅源, 直接与R和F混合形成溶胶, 凝胶并超临界干燥后获得RF/SiO2 复合气凝胶[15 ,24 -25 ] .APTES含量的增加将减少混合溶胶体系的凝胶时间[24 ,27 ] .在该溶胶体系中, APTES除了作为硅源先驱体参与水解缩聚反应形成Si-O-Si网络结构以外, 还可作为“内部催化剂(internal catalyst)”促进R和F之间的缩聚反应.这是由于APTES中氨基的水合作用(hydration of amino groups)在APTES水解产物中形成了环状的分子间氢键, 为反应提供了良好的通路, 从而加速溶胶-凝胶过程[27 ] .RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
... [26 ].APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
... [26 ].除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
Aminopolysiloxane gels: production and properties.
2
1996
... 为了避免以TEOS或正硅酸甲酯(tetramethy lorthosilicate, TMOS)为硅源制备RF/SiO2 气凝胶时的复杂溶胶-凝胶过程, 进一步减少水解缩聚的时间, 有研究采用硅烷偶联剂作为硅源一步合成RF/ SiO2 气凝胶[15 ,24 -26 ] .在该类方法中, 硅烷偶联剂3-氨基丙基三乙氧基硅烷[(3-aminopropyl)triethoxysilane, APTES]可作为硅源, 直接与R和F混合形成溶胶, 凝胶并超临界干燥后获得RF/SiO2 复合气凝胶[15 ,24 -25 ] .APTES含量的增加将减少混合溶胶体系的凝胶时间[24 ,27 ] .在该溶胶体系中, APTES除了作为硅源先驱体参与水解缩聚反应形成Si-O-Si网络结构以外, 还可作为“内部催化剂(internal catalyst)”促进R和F之间的缩聚反应.这是由于APTES中氨基的水合作用(hydration of amino groups)在APTES水解产物中形成了环状的分子间氢键, 为反应提供了良好的通路, 从而加速溶胶-凝胶过程[27 ] .RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
... [27 ].RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
Role of pores in the carbothermal reduction of carbon-silica nanocomposites into silicon carbide nanostructures.
5
2007
... 为了避免以TEOS或正硅酸甲酯(tetramethy lorthosilicate, TMOS)为硅源制备RF/SiO2 气凝胶时的复杂溶胶-凝胶过程, 进一步减少水解缩聚的时间, 有研究采用硅烷偶联剂作为硅源一步合成RF/ SiO2 气凝胶[15 ,24 -26 ] .在该类方法中, 硅烷偶联剂3-氨基丙基三乙氧基硅烷[(3-aminopropyl)triethoxysilane, APTES]可作为硅源, 直接与R和F混合形成溶胶, 凝胶并超临界干燥后获得RF/SiO2 复合气凝胶[15 ,24 -25 ] .APTES含量的增加将减少混合溶胶体系的凝胶时间[24 ,27 ] .在该溶胶体系中, APTES除了作为硅源先驱体参与水解缩聚反应形成Si-O-Si网络结构以外, 还可作为“内部催化剂(internal catalyst)”促进R和F之间的缩聚反应.这是由于APTES中氨基的水合作用(hydration of amino groups)在APTES水解产物中形成了环状的分子间氢键, 为反应提供了良好的通路, 从而加速溶胶-凝胶过程[27 ] .RF/SiO2 气凝胶经高温烧结转变成C/SiO2 气凝胶后(高APTES含量时复合材料的骨架主要由SiO2 构成; 低APTES含量时骨架则主要由C构成[15 ] ), 经1500℃处理转变为SiC气凝胶后, 在650℃空气环境下具有良好的抗氧化能力[25 ] .以APTES和TEOS作为混合硅源先驱体合成的RF/SiO2 气凝胶, APTES在促进溶胶-凝胶反应过程的同时, 还增大了样品的密度.经热处理后, 由于APTES中氨丙基(aminopropyl)基团的存在, 引起RF/SiO2 气凝胶有更多的质量损失和体积收缩[26 ] .APTES中的Si-C键加速了碳热还原反应的进行, 导致α-SiC的形成[26 ] .除此以外, 若用NaOH溶液将C/SiO2 气凝胶中的SiO2 蚀刻去掉, 则可获得高比表面积的碳气凝胶[28 ] . ...
... 在这一过程中, SiC首先在先驱体表面形核(nuclei), 随后长大成纳米晶须(nanowhiskers).其间相互贯穿孔的存在为气体产物(如SiO、CO和CO2 )的扩散提供了通路, 加速了这一反应的进行[12 ] .C与SiO2 的比例和C/SiO2 复合材料的致密度是影响SiC反应动力学和最终SiC微观结构的重要因素.图3 给出了不同结构SiC的形成过程示意图[28 ] .高碳硅比例下, 介孔C/SiO2 可形成纳米级纤维状和颗粒状SiC, 如图3 (a)所示; 低碳硅比例下, 除形成纳米纤维状和颗粒状SiC以外, 还渗透着少量自由C, 如图3 (b)所示.其中, 纤维状和颗粒状SiC产物的比例可通过改变碳硅间的比例并利用C的渗透加以调节.高碳硅比例的致密C/SiO2 可转变为介孔SiC, 如图3 (c)所示.这是由于高碳硅比例以及C/SiO2 和C之间界面结构可以提高碳热还原反应速率, 介孔在为气态产物提供扩散通道的同时, 也会限制介孔C/SiO2 材料中SiC的生长[28 ] .除此以外, 采用低温镁热反应(low-temperature magnesiothermic reaction)可使C/SiO2 材料在700℃的低温下转变成SiC[23 ] .通过碳热还原RF/SiO2 气凝胶制得的材料是以β- SiC为主体, 并含有少量α-SiC的SiC或C/SiC气凝胶材料.该材料具有多孔结构和高的比表面积, 并具有高硬度、好的抗热震能力、高热导和高稳定性、低膨胀系数和大的带隙, 可用于增强相、催化剂、高能和高频电子材料、光电材料、抗辐射材料、氢分离膜载体和吸波器件等[12 ] . ...
... [28 ].除此以外, 采用低温镁热反应(low-temperature magnesiothermic reaction)可使C/SiO2 材料在700℃的低温下转变成SiC[23 ] .通过碳热还原RF/SiO2 气凝胶制得的材料是以β- SiC为主体, 并含有少量α-SiC的SiC或C/SiC气凝胶材料.该材料具有多孔结构和高的比表面积, 并具有高硬度、好的抗热震能力、高热导和高稳定性、低膨胀系数和大的带隙, 可用于增强相、催化剂、高能和高频电子材料、光电材料、抗辐射材料、氢分离膜载体和吸波器件等[12 ] . ...
... SiC形成过程示意图[28 ] ...
... Schematic illustration of SiC formation[28 ] ...
Nanoporous phloroglucinol- formaldehyde carbon aerogels for electrochemical use.
1
2005
... 采用共聚法制备C/SiO2 和C/SiC气凝胶, 除了以R和F的聚合物为先驱体之外, 碳的来源还可能是间苯三酚-F(Phloroglucinol-Formaldehyde, PF)[29 ] 、可溶性酚醛树脂(resol)[30 ] 、苯酚-R-F(phenol-resorcinol- formaldehyde, PRF)[31 ] 、蔗糖(saccharose)[32 ] 、淀粉(starch)[33 ] 、石油焦(petroleum coke)[34 ] 、煤沥青(coal tar pitch)[35 ] 、酚醛清漆(novolac)[36 ] 等.从经济角度来看, 以经济衍生物作为碳源可以有效地改善R作为原料时价格高的缺点, 但这种碳源成分较复杂.而以蔗糖、淀粉、石油焦等作为碳源很难能形成交联的网络结构, 无法得到高孔隙率的块状气凝胶材料. ...
Stabilizing surfactant templated cylindrical mesopores in polymer and carbon films through composite formation with silica reinforcement.
1
2010
... 采用共聚法制备C/SiO2 和C/SiC气凝胶, 除了以R和F的聚合物为先驱体之外, 碳的来源还可能是间苯三酚-F(Phloroglucinol-Formaldehyde, PF)[29 ] 、可溶性酚醛树脂(resol)[30 ] 、苯酚-R-F(phenol-resorcinol- formaldehyde, PRF)[31 ] 、蔗糖(saccharose)[32 ] 、淀粉(starch)[33 ] 、石油焦(petroleum coke)[34 ] 、煤沥青(coal tar pitch)[35 ] 、酚醛清漆(novolac)[36 ] 等.从经济角度来看, 以经济衍生物作为碳源可以有效地改善R作为原料时价格高的缺点, 但这种碳源成分较复杂.而以蔗糖、淀粉、石油焦等作为碳源很难能形成交联的网络结构, 无法得到高孔隙率的块状气凝胶材料. ...
Synthesis of porous silicon nitride using silica/carbon composite derived from phenol-resorcinol-formaldehyde gel.
1
2016
... 采用共聚法制备C/SiO2 和C/SiC气凝胶, 除了以R和F的聚合物为先驱体之外, 碳的来源还可能是间苯三酚-F(Phloroglucinol-Formaldehyde, PF)[29 ] 、可溶性酚醛树脂(resol)[30 ] 、苯酚-R-F(phenol-resorcinol- formaldehyde, PRF)[31 ] 、蔗糖(saccharose)[32 ] 、淀粉(starch)[33 ] 、石油焦(petroleum coke)[34 ] 、煤沥青(coal tar pitch)[35 ] 、酚醛清漆(novolac)[36 ] 等.从经济角度来看, 以经济衍生物作为碳源可以有效地改善R作为原料时价格高的缺点, 但这种碳源成分较复杂.而以蔗糖、淀粉、石油焦等作为碳源很难能形成交联的网络结构, 无法得到高孔隙率的块状气凝胶材料. ...
Preparations of C/SiC composites and their use as supports for Ru catalyst in ammonia synthesis.
1
2009
... 采用共聚法制备C/SiO2 和C/SiC气凝胶, 除了以R和F的聚合物为先驱体之外, 碳的来源还可能是间苯三酚-F(Phloroglucinol-Formaldehyde, PF)[29 ] 、可溶性酚醛树脂(resol)[30 ] 、苯酚-R-F(phenol-resorcinol- formaldehyde, PRF)[31 ] 、蔗糖(saccharose)[32 ] 、淀粉(starch)[33 ] 、石油焦(petroleum coke)[34 ] 、煤沥青(coal tar pitch)[35 ] 、酚醛清漆(novolac)[36 ] 等.从经济角度来看, 以经济衍生物作为碳源可以有效地改善R作为原料时价格高的缺点, 但这种碳源成分较复杂.而以蔗糖、淀粉、石油焦等作为碳源很难能形成交联的网络结构, 无法得到高孔隙率的块状气凝胶材料. ...
Synthesis of silicon carbide through the sol-gel process from different precursors.
1
1995
... 采用共聚法制备C/SiO2 和C/SiC气凝胶, 除了以R和F的聚合物为先驱体之外, 碳的来源还可能是间苯三酚-F(Phloroglucinol-Formaldehyde, PF)[29 ] 、可溶性酚醛树脂(resol)[30 ] 、苯酚-R-F(phenol-resorcinol- formaldehyde, PRF)[31 ] 、蔗糖(saccharose)[32 ] 、淀粉(starch)[33 ] 、石油焦(petroleum coke)[34 ] 、煤沥青(coal tar pitch)[35 ] 、酚醛清漆(novolac)[36 ] 等.从经济角度来看, 以经济衍生物作为碳源可以有效地改善R作为原料时价格高的缺点, 但这种碳源成分较复杂.而以蔗糖、淀粉、石油焦等作为碳源很难能形成交联的网络结构, 无法得到高孔隙率的块状气凝胶材料. ...
Synthesis of nanometre silicon carbide whiskers from binary carbonaceous silica aerogels.
1
2001
... 采用共聚法制备C/SiO2 和C/SiC气凝胶, 除了以R和F的聚合物为先驱体之外, 碳的来源还可能是间苯三酚-F(Phloroglucinol-Formaldehyde, PF)[29 ] 、可溶性酚醛树脂(resol)[30 ] 、苯酚-R-F(phenol-resorcinol- formaldehyde, PRF)[31 ] 、蔗糖(saccharose)[32 ] 、淀粉(starch)[33 ] 、石油焦(petroleum coke)[34 ] 、煤沥青(coal tar pitch)[35 ] 、酚醛清漆(novolac)[36 ] 等.从经济角度来看, 以经济衍生物作为碳源可以有效地改善R作为原料时价格高的缺点, 但这种碳源成分较复杂.而以蔗糖、淀粉、石油焦等作为碳源很难能形成交联的网络结构, 无法得到高孔隙率的块状气凝胶材料. ...
Formation of black glasses and silicon carbide from binary carbonaceous/silica hydrogels.
1
1995
... 采用共聚法制备C/SiO2 和C/SiC气凝胶, 除了以R和F的聚合物为先驱体之外, 碳的来源还可能是间苯三酚-F(Phloroglucinol-Formaldehyde, PF)[29 ] 、可溶性酚醛树脂(resol)[30 ] 、苯酚-R-F(phenol-resorcinol- formaldehyde, PRF)[31 ] 、蔗糖(saccharose)[32 ] 、淀粉(starch)[33 ] 、石油焦(petroleum coke)[34 ] 、煤沥青(coal tar pitch)[35 ] 、酚醛清漆(novolac)[36 ] 等.从经济角度来看, 以经济衍生物作为碳源可以有效地改善R作为原料时价格高的缺点, 但这种碳源成分较复杂.而以蔗糖、淀粉、石油焦等作为碳源很难能形成交联的网络结构, 无法得到高孔隙率的块状气凝胶材料. ...
Preparation and characterization of C/SiO2 /SiC aerogels based on novolac/silica hybrid hyperporous materials.
1
2015
... 采用共聚法制备C/SiO2 和C/SiC气凝胶, 除了以R和F的聚合物为先驱体之外, 碳的来源还可能是间苯三酚-F(Phloroglucinol-Formaldehyde, PF)[29 ] 、可溶性酚醛树脂(resol)[30 ] 、苯酚-R-F(phenol-resorcinol- formaldehyde, PRF)[31 ] 、蔗糖(saccharose)[32 ] 、淀粉(starch)[33 ] 、石油焦(petroleum coke)[34 ] 、煤沥青(coal tar pitch)[35 ] 、酚醛清漆(novolac)[36 ] 等.从经济角度来看, 以经济衍生物作为碳源可以有效地改善R作为原料时价格高的缺点, 但这种碳源成分较复杂.而以蔗糖、淀粉、石油焦等作为碳源很难能形成交联的网络结构, 无法得到高孔隙率的块状气凝胶材料. ...
Heavy metals removal using activated carbon, silica and silica activated carbon composite.
1
2014
... 在吸附领域, 活性炭经SiO2 溶胶改性后, 借助于SiO2 气凝胶纳米多孔的结构特性, 可以提高活性炭材料的比表面积和孔隙率, 进而提高活性炭的吸附率.Zhou等[10 ] 将活性炭浸入以TEOS和苯基三乙氧基硅烷(phenyltriethoxysilane, PTES)或甲基三乙氧基硅烷(methyltriethoxysilane, MTES)为混合先驱体制备的SiO2 溶胶中, SiO2 溶胶颗粒原位聚合于碳材料表面, 获得了SiO2 /活性炭复合材料.该材料的微观结构上是SiO2 气凝胶包裹活性炭颗粒, 且骨架结构比较松散.依靠表面吸附(surface adsorption)作用, 将其吸附率从活性炭的76%提高到96.5%.该材料可实现从废水中吸附三硝基甲苯(2,4,6-trinitrotoluene, TNT)的作用.同时, 有机硅氧烷先驱体提供的不同疏水基团对吸附率也有影响.Karnib等[37 ] 用APTES对SiO2 颗粒进行表面修饰后, 与活性炭在溶液状态下混合, 经干燥和研磨后, 获得的SiO2 /活性炭复合颗粒对水溶液中的重金属离子具有很好的吸附效果. ...
ARDUINI-SCHUSTER M C, et al. Thermal transport in organic and opacified silica monolithic aerogels.
1
1992
... 在隔热领域, 由于碳材料对红外波段具有良好的吸收特性, 通常引入SiO2 气凝胶提高其高温抗辐射能力.Lu等[38 ] 将炭黑作为红外遮光剂分散在SiO2 气凝胶中, 可以明显降低高温辐射热传导.Liu等[39 ] 将碳泡沫浸入SiO2 溶胶中, 凝胶并干燥后形成了碳泡沫/ SiO2 气凝胶复合材料.由于碳泡沫表面被SiO2 气凝胶覆盖形成核壳结构, 该复合材料的热导率比纯碳泡沫降低了41.9 %, 但其压缩强度并没有明显下降. ...
Thermal insulation composite prepared from carbon foam and silica aerogel under ambient pressure.
1
2015
... 在隔热领域, 由于碳材料对红外波段具有良好的吸收特性, 通常引入SiO2 气凝胶提高其高温抗辐射能力.Lu等[38 ] 将炭黑作为红外遮光剂分散在SiO2 气凝胶中, 可以明显降低高温辐射热传导.Liu等[39 ] 将碳泡沫浸入SiO2 溶胶中, 凝胶并干燥后形成了碳泡沫/ SiO2 气凝胶复合材料.由于碳泡沫表面被SiO2 气凝胶覆盖形成核壳结构, 该复合材料的热导率比纯碳泡沫降低了41.9 %, 但其压缩强度并没有明显下降. ...
Improved thermal stability, properties, and electrocatalytic activity of Sol-Gel silica modified carbon supported Pt catalysts.
1
2012
... 在电催化方面, 经SiO2 修饰的碳载铂催化剂(carbon-supported platinum catalysts), 其热稳定性和电催化特性得以提高, 可用于低温质子交换膜(proton exchange membrane, PEM)燃料电池的电催化领域[40 ] . ...
Micromechanical properties of carbon-silica aerogel composites.
1
2002
... 除此以外, 在SiO2 气凝胶中填充炭粉, 可增强材料的弹性回复能力, 并保持SiO2 气凝胶基体的硬度, 这有助于减小气凝胶在超临界干燥过程中的收缩[41 ] , 并可获得更高比表面积的C/SiO2 复合材料[42 ] .C和SiO2 在C/SiO2 复合材料中的相对分布状态与各自在溶液中的等电点(isoelectric points, IEPs)有关[43 ] .Spassova等[43 ] 研究发现, 与SiO2 进行复合时, 如果不同形态碳材料的IEPs高于SiO2 的IEPs(pH=2.5), 则基于静电反应(electrostatic interaction)形成以SiO2 为核、C为壳层的结构形式; 如果碳材料的IEPs低于SiO2 的IEPs, 则将出现C颗粒表面吸附Si-OH的结构形式.经SiO2 溶胶浸入后的多孔碳材料(如碳气凝胶), 由于碳骨架表面包裹了一层耐氧化的SiO2 凝胶, 可以提高碳材料的热稳定性[44 ] .同时, Worsley等[44 ] 发现将比表面积高达3000 m2 /g的碳气凝胶浸入SiO2 溶胶后, 由于纤细的碳骨架表面均匀覆盖着一层SiO2 凝胶, 为C与SiO2 之间的充分化学反应创造了条件.因而, 经1500℃碳热还原反应后, C/SiO2 复合气凝胶材料可转化为比表面积达2000 m2 /g的SiC多孔材料.这种方法为形成高比表面积的金属氧化物、碳化物或氮化物提供了一种好的研究思路.另外, C/SiO2 复合材料在高温下向SiC转变的过程中, 由共聚法获得的C/SiO2 气凝胶转变成SiC时, 常以纳米晶须状(whiskerlike)而不是颗粒状(particulate)的形式存在, 但这一晶须状结构通常并不是理想的SiC材料形式.为了克服这一问题, 通常需要在SiO2 表面包覆C的先驱体, 即形成C包SiO2 的胶囊式结构, 以确保所有在SiO2 和C界面间产生的气态SiO与C发生反应, 进而形成颗粒状SiC材料[45 ] .图4 给出了SiO2 与C界面间SiC形成的过程示意图[45 ] . ...
Carbon-silica xerogel and aerogel composites.
3
1995
... 除此以外, 在SiO2 气凝胶中填充炭粉, 可增强材料的弹性回复能力, 并保持SiO2 气凝胶基体的硬度, 这有助于减小气凝胶在超临界干燥过程中的收缩[41 ] , 并可获得更高比表面积的C/SiO2 复合材料[42 ] .C和SiO2 在C/SiO2 复合材料中的相对分布状态与各自在溶液中的等电点(isoelectric points, IEPs)有关[43 ] .Spassova等[43 ] 研究发现, 与SiO2 进行复合时, 如果不同形态碳材料的IEPs高于SiO2 的IEPs(pH=2.5), 则基于静电反应(electrostatic interaction)形成以SiO2 为核、C为壳层的结构形式; 如果碳材料的IEPs低于SiO2 的IEPs, 则将出现C颗粒表面吸附Si-OH的结构形式.经SiO2 溶胶浸入后的多孔碳材料(如碳气凝胶), 由于碳骨架表面包裹了一层耐氧化的SiO2 凝胶, 可以提高碳材料的热稳定性[44 ] .同时, Worsley等[44 ] 发现将比表面积高达3000 m2 /g的碳气凝胶浸入SiO2 溶胶后, 由于纤细的碳骨架表面均匀覆盖着一层SiO2 凝胶, 为C与SiO2 之间的充分化学反应创造了条件.因而, 经1500℃碳热还原反应后, C/SiO2 复合气凝胶材料可转化为比表面积达2000 m2 /g的SiC多孔材料.这种方法为形成高比表面积的金属氧化物、碳化物或氮化物提供了一种好的研究思路.另外, C/SiO2 复合材料在高温下向SiC转变的过程中, 由共聚法获得的C/SiO2 气凝胶转变成SiC时, 常以纳米晶须状(whiskerlike)而不是颗粒状(particulate)的形式存在, 但这一晶须状结构通常并不是理想的SiC材料形式.为了克服这一问题, 通常需要在SiO2 表面包覆C的先驱体, 即形成C包SiO2 的胶囊式结构, 以确保所有在SiO2 和C界面间产生的气态SiO与C发生反应, 进而形成颗粒状SiC材料[45 ] .图4 给出了SiO2 与C界面间SiC形成的过程示意图[45 ] . ...
... 通常, 烃基取代硅氧烷先驱体在水解缩聚过程中, 由于不可水解R基团的存在, 往往对硅烷醇的缩聚造成阻碍, 从而使其难以凝胶.因此, 在溶胶-凝胶阶段常借助易于水解缩聚的四烷氧基硅烷(如TMOS、TEOS)构成交联网络结构.Babonneau等[50 ] 采用二甲基二乙氧基硅烷(dimethyldiethoxysilane, DEDMS)和TEOS作为双先驱体, 经水解缩聚后形成了Si-C-O凝胶体系.29 Si核磁共振(nuclear magnetic resonance, NMR)证明这两种双先驱体之间发生了共聚合化作用, 并存在两类凝胶单元, 即源于DEDMS构成的(CH3 )2 Si(O0.5 )2 和源于TEOS构成的Si(O0.5 )4 .Liu等[42 ] 采用苯基三甲氧基硅烷(phenyltrimethoxysilane, PhTMS)和TMOS作为双先驱体制备的凝胶体系由O3/2 -Si-C6 H5 和1/2 O-Si-O1/2 凝胶单元构成.随着PhTMS含量的增加, 孔大小、孔体积和比表面积均下降, 经1000℃热解后, 获得的SiCO仍旧保持 581 m2 /g的高比表面积.Feng等[51 ] 以TEOS和聚二甲基硅氧烷(polydimethylsiloxane, PDMS)为双先驱体, 经溶胶-凝胶、超临界干燥和1200℃热解后得到块状SiCO气凝胶单体.该材料呈非晶态, 其网络结构可在1100℃下保持稳定, 当温度达到1200℃, 非晶态中逐渐开始出现晶相, 而此时仅有1.65%的质量损失, 可见该材料具有很高的热稳定性.另外, 该材料的密度为0.3 g/cm3 , 热导率仅为0.027 W/(m·K), 可作为一种高温隔热材料使用. ...
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
Impact of carbon on the surface and activity of silica-carbon supported copper catalysts for reduction of nitrogen oxides.
2
2016
... 除此以外, 在SiO2 气凝胶中填充炭粉, 可增强材料的弹性回复能力, 并保持SiO2 气凝胶基体的硬度, 这有助于减小气凝胶在超临界干燥过程中的收缩[41 ] , 并可获得更高比表面积的C/SiO2 复合材料[42 ] .C和SiO2 在C/SiO2 复合材料中的相对分布状态与各自在溶液中的等电点(isoelectric points, IEPs)有关[43 ] .Spassova等[43 ] 研究发现, 与SiO2 进行复合时, 如果不同形态碳材料的IEPs高于SiO2 的IEPs(pH=2.5), 则基于静电反应(electrostatic interaction)形成以SiO2 为核、C为壳层的结构形式; 如果碳材料的IEPs低于SiO2 的IEPs, 则将出现C颗粒表面吸附Si-OH的结构形式.经SiO2 溶胶浸入后的多孔碳材料(如碳气凝胶), 由于碳骨架表面包裹了一层耐氧化的SiO2 凝胶, 可以提高碳材料的热稳定性[44 ] .同时, Worsley等[44 ] 发现将比表面积高达3000 m2 /g的碳气凝胶浸入SiO2 溶胶后, 由于纤细的碳骨架表面均匀覆盖着一层SiO2 凝胶, 为C与SiO2 之间的充分化学反应创造了条件.因而, 经1500℃碳热还原反应后, C/SiO2 复合气凝胶材料可转化为比表面积达2000 m2 /g的SiC多孔材料.这种方法为形成高比表面积的金属氧化物、碳化物或氮化物提供了一种好的研究思路.另外, C/SiO2 复合材料在高温下向SiC转变的过程中, 由共聚法获得的C/SiO2 气凝胶转变成SiC时, 常以纳米晶须状(whiskerlike)而不是颗粒状(particulate)的形式存在, 但这一晶须状结构通常并不是理想的SiC材料形式.为了克服这一问题, 通常需要在SiO2 表面包覆C的先驱体, 即形成C包SiO2 的胶囊式结构, 以确保所有在SiO2 和C界面间产生的气态SiO与C发生反应, 进而形成颗粒状SiC材料[45 ] .图4 给出了SiO2 与C界面间SiC形成的过程示意图[45 ] . ...
... [43 ]研究发现, 与SiO2 进行复合时, 如果不同形态碳材料的IEPs高于SiO2 的IEPs(pH=2.5), 则基于静电反应(electrostatic interaction)形成以SiO2 为核、C为壳层的结构形式; 如果碳材料的IEPs低于SiO2 的IEPs, 则将出现C颗粒表面吸附Si-OH的结构形式.经SiO2 溶胶浸入后的多孔碳材料(如碳气凝胶), 由于碳骨架表面包裹了一层耐氧化的SiO2 凝胶, 可以提高碳材料的热稳定性[44 ] .同时, Worsley等[44 ] 发现将比表面积高达3000 m2 /g的碳气凝胶浸入SiO2 溶胶后, 由于纤细的碳骨架表面均匀覆盖着一层SiO2 凝胶, 为C与SiO2 之间的充分化学反应创造了条件.因而, 经1500℃碳热还原反应后, C/SiO2 复合气凝胶材料可转化为比表面积达2000 m2 /g的SiC多孔材料.这种方法为形成高比表面积的金属氧化物、碳化物或氮化物提供了一种好的研究思路.另外, C/SiO2 复合材料在高温下向SiC转变的过程中, 由共聚法获得的C/SiO2 气凝胶转变成SiC时, 常以纳米晶须状(whiskerlike)而不是颗粒状(particulate)的形式存在, 但这一晶须状结构通常并不是理想的SiC材料形式.为了克服这一问题, 通常需要在SiO2 表面包覆C的先驱体, 即形成C包SiO2 的胶囊式结构, 以确保所有在SiO2 和C界面间产生的气态SiO与C发生反应, 进而形成颗粒状SiC材料[45 ] .图4 给出了SiO2 与C界面间SiC形成的过程示意图[45 ] . ...
Synthesis and characterization of monolithic, high surface area SiO2 /C and SiC/C composites.
2
2010
... 除此以外, 在SiO2 气凝胶中填充炭粉, 可增强材料的弹性回复能力, 并保持SiO2 气凝胶基体的硬度, 这有助于减小气凝胶在超临界干燥过程中的收缩[41 ] , 并可获得更高比表面积的C/SiO2 复合材料[42 ] .C和SiO2 在C/SiO2 复合材料中的相对分布状态与各自在溶液中的等电点(isoelectric points, IEPs)有关[43 ] .Spassova等[43 ] 研究发现, 与SiO2 进行复合时, 如果不同形态碳材料的IEPs高于SiO2 的IEPs(pH=2.5), 则基于静电反应(electrostatic interaction)形成以SiO2 为核、C为壳层的结构形式; 如果碳材料的IEPs低于SiO2 的IEPs, 则将出现C颗粒表面吸附Si-OH的结构形式.经SiO2 溶胶浸入后的多孔碳材料(如碳气凝胶), 由于碳骨架表面包裹了一层耐氧化的SiO2 凝胶, 可以提高碳材料的热稳定性[44 ] .同时, Worsley等[44 ] 发现将比表面积高达3000 m2 /g的碳气凝胶浸入SiO2 溶胶后, 由于纤细的碳骨架表面均匀覆盖着一层SiO2 凝胶, 为C与SiO2 之间的充分化学反应创造了条件.因而, 经1500℃碳热还原反应后, C/SiO2 复合气凝胶材料可转化为比表面积达2000 m2 /g的SiC多孔材料.这种方法为形成高比表面积的金属氧化物、碳化物或氮化物提供了一种好的研究思路.另外, C/SiO2 复合材料在高温下向SiC转变的过程中, 由共聚法获得的C/SiO2 气凝胶转变成SiC时, 常以纳米晶须状(whiskerlike)而不是颗粒状(particulate)的形式存在, 但这一晶须状结构通常并不是理想的SiC材料形式.为了克服这一问题, 通常需要在SiO2 表面包覆C的先驱体, 即形成C包SiO2 的胶囊式结构, 以确保所有在SiO2 和C界面间产生的气态SiO与C发生反应, 进而形成颗粒状SiC材料[45 ] .图4 给出了SiO2 与C界面间SiC形成的过程示意图[45 ] . ...
... [44 ]发现将比表面积高达3000 m2 /g的碳气凝胶浸入SiO2 溶胶后, 由于纤细的碳骨架表面均匀覆盖着一层SiO2 凝胶, 为C与SiO2 之间的充分化学反应创造了条件.因而, 经1500℃碳热还原反应后, C/SiO2 复合气凝胶材料可转化为比表面积达2000 m2 /g的SiC多孔材料.这种方法为形成高比表面积的金属氧化物、碳化物或氮化物提供了一种好的研究思路.另外, C/SiO2 复合材料在高温下向SiC转变的过程中, 由共聚法获得的C/SiO2 气凝胶转变成SiC时, 常以纳米晶须状(whiskerlike)而不是颗粒状(particulate)的形式存在, 但这一晶须状结构通常并不是理想的SiC材料形式.为了克服这一问题, 通常需要在SiO2 表面包覆C的先驱体, 即形成C包SiO2 的胶囊式结构, 以确保所有在SiO2 和C界面间产生的气态SiO与C发生反应, 进而形成颗粒状SiC材料[45 ] .图4 给出了SiO2 与C界面间SiC形成的过程示意图[45 ] . ...
Click synthesis of monolithic silicon carbide aerogels from polyacrylonitrile-coated 3D silica networks.
4
2010
... 除此以外, 在SiO2 气凝胶中填充炭粉, 可增强材料的弹性回复能力, 并保持SiO2 气凝胶基体的硬度, 这有助于减小气凝胶在超临界干燥过程中的收缩[41 ] , 并可获得更高比表面积的C/SiO2 复合材料[42 ] .C和SiO2 在C/SiO2 复合材料中的相对分布状态与各自在溶液中的等电点(isoelectric points, IEPs)有关[43 ] .Spassova等[43 ] 研究发现, 与SiO2 进行复合时, 如果不同形态碳材料的IEPs高于SiO2 的IEPs(pH=2.5), 则基于静电反应(electrostatic interaction)形成以SiO2 为核、C为壳层的结构形式; 如果碳材料的IEPs低于SiO2 的IEPs, 则将出现C颗粒表面吸附Si-OH的结构形式.经SiO2 溶胶浸入后的多孔碳材料(如碳气凝胶), 由于碳骨架表面包裹了一层耐氧化的SiO2 凝胶, 可以提高碳材料的热稳定性[44 ] .同时, Worsley等[44 ] 发现将比表面积高达3000 m2 /g的碳气凝胶浸入SiO2 溶胶后, 由于纤细的碳骨架表面均匀覆盖着一层SiO2 凝胶, 为C与SiO2 之间的充分化学反应创造了条件.因而, 经1500℃碳热还原反应后, C/SiO2 复合气凝胶材料可转化为比表面积达2000 m2 /g的SiC多孔材料.这种方法为形成高比表面积的金属氧化物、碳化物或氮化物提供了一种好的研究思路.另外, C/SiO2 复合材料在高温下向SiC转变的过程中, 由共聚法获得的C/SiO2 气凝胶转变成SiC时, 常以纳米晶须状(whiskerlike)而不是颗粒状(particulate)的形式存在, 但这一晶须状结构通常并不是理想的SiC材料形式.为了克服这一问题, 通常需要在SiO2 表面包覆C的先驱体, 即形成C包SiO2 的胶囊式结构, 以确保所有在SiO2 和C界面间产生的气态SiO与C发生反应, 进而形成颗粒状SiC材料[45 ] .图4 给出了SiO2 与C界面间SiC形成的过程示意图[45 ] . ...
... [45 ]. ...
... SiO2 与C界面间SiC形成的过程示意图[45 ] ...
... Processes of formed silicon carbide at the interface between silica and carbon[45 ] ...
ZHANGH. Silicon oxycarbide glasses.
4
1999
... 聚合物先驱体热解法是指以溶胶-凝胶法形成的有机硅氧烷聚合物凝胶经高温热解转变为SiCO (silicon oxycarbide)或SiC材料的方法.SiCO具有C和O同时与Si相结合的化学结构特征.这一四面体网络结构可定义为Cx 4- x x =1, 2或3)[46 ] .相较于O2- 仅可提供两个阴离子配位键而言, C取代O后, 有提供3到4个阴离子配位的可能性, 这使得SiCO比纯SiO2 具有更高的交联度[47 ] .基于该方法制备的SiCO, 常具有耐高温、轻质、耐腐蚀、低热导等性能特点, 在高温传感器、催化、过滤、电池电极材料以及高温热防护等领域具有广泛的应用前景. ...
... 采用溶胶-凝胶法合成SiCO是一种最佳的制备方法.为了在这一过程中均匀地引入Si-C键, 常以带有直接与Si原子相连烃基的有机硅氧烷作为先驱体, 即烃基取代硅氧烷先驱体(alkyl-substituted silicon-alkoxide precursors)[46 ] .该先驱体的分子式可表示为Rx 4- x x 可取1~3), 其中, R既可为饱和烃基(如CH3 、C2 H5 ), 也可为不饱和烃基(如C6 H5 ); R’主要为CH3 或C2 H5 [48 ] .在溶胶-凝胶过程中, 与Si直接相连的R并不参与水解, 因而可保留在以Si-O-Si为主体构成的网络结构中.R中含有的碳链长短和数量, 将决定SiCO中Si-C键的数量, 因此, 先驱体的分子组成是影响SiCO最终构成的最直接因素.有研究表明[49 ] , 凝胶中C含量会随着R基团中碳链长度的增加而呈比例增加.但是, 热解后保留的C含量与先驱体或凝胶中存在的碳链长度并无明确比例关系.随着R基团中C含量的降低, C-Si-O键所占比例将增加.热解后, 只有直接与Si原子相连的C原子才可以SiCO的形式保留在材料中, 而其他C原子则以自由C的形式存在于材料中. ...
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
... 经热解法制备的SiCO比SiO2 具有更高的力学、热学和化学稳定性[46 ] .在SiCO中, C以自由碳或以与Si连接形成SiC4- x x 4 基团的形式存在.相比于硅氧间只能形成二氧连接, 硅碳间最多的四碳连接形式则可形成更为坚固的刚性骨架, 从而提高SiCO的模量、硬度、粘度、密度和玻璃转化温度.当体系中的自由C含量逐渐升高时, SiCO的弹性模量、弯曲强度、维氏硬度都会出现明显增加[63 -65 ] .表2 比较了SiCO和玻璃态SiO2 之间的物理特性[64 ] .在化学稳定性方面, SiCO经历如碱性条件或HF的腐蚀后, 由于其中的Si-C键可经受更高的亲核攻击(nucleophilic attack), 具有更高的无序和交联度, 并且与Si原子相连的C和自由C均可对腐蚀反应起阻碍作用, 因而比SiO2 表现出更好的化学稳定性[60 ] .针对多孔SiCO的传热问题, Qiu等[66 ] 建立了一种描述其热传递的立方阵列交叉纳米球模型(cubic array of an intersecting nano-spheres model), 并发展了预测有效热导率的理论公式.结果表明, 多孔SiCO室温下的有效热导率在0.041~0.078 W/(m·K)之间, 该数值主要与SiCO固相体积分数以及相邻颗粒间接触长度(a )与颗粒直径(d )的比值a /d 有关.图5 给出了该研究中建立的多孔SiCO陶瓷的几何结构和热传递分析模型[66 ] . ...
High surface area SiC/silicon oxycarbide glasses prepared from phenyltrimethoxysilane- tetramethoxysilane gels.
1
1996
... 聚合物先驱体热解法是指以溶胶-凝胶法形成的有机硅氧烷聚合物凝胶经高温热解转变为SiCO (silicon oxycarbide)或SiC材料的方法.SiCO具有C和O同时与Si相结合的化学结构特征.这一四面体网络结构可定义为Cx 4- x x =1, 2或3)[46 ] .相较于O2- 仅可提供两个阴离子配位键而言, C取代O后, 有提供3到4个阴离子配位的可能性, 这使得SiCO比纯SiO2 具有更高的交联度[47 ] .基于该方法制备的SiCO, 常具有耐高温、轻质、耐腐蚀、低热导等性能特点, 在高温传感器、催化、过滤、电池电极材料以及高温热防护等领域具有广泛的应用前景. ...
Porous silicon oxycarbide glasses.
2
1996
... 采用溶胶-凝胶法合成SiCO是一种最佳的制备方法.为了在这一过程中均匀地引入Si-C键, 常以带有直接与Si原子相连烃基的有机硅氧烷作为先驱体, 即烃基取代硅氧烷先驱体(alkyl-substituted silicon-alkoxide precursors)[46 ] .该先驱体的分子式可表示为Rx 4- x x 可取1~3), 其中, R既可为饱和烃基(如CH3 、C2 H5 ), 也可为不饱和烃基(如C6 H5 ); R’主要为CH3 或C2 H5 [48 ] .在溶胶-凝胶过程中, 与Si直接相连的R并不参与水解, 因而可保留在以Si-O-Si为主体构成的网络结构中.R中含有的碳链长短和数量, 将决定SiCO中Si-C键的数量, 因此, 先驱体的分子组成是影响SiCO最终构成的最直接因素.有研究表明[49 ] , 凝胶中C含量会随着R基团中碳链长度的增加而呈比例增加.但是, 热解后保留的C含量与先驱体或凝胶中存在的碳链长度并无明确比例关系.随着R基团中C含量的降低, C-Si-O键所占比例将增加.热解后, 只有直接与Si原子相连的C原子才可以SiCO的形式保留在材料中, 而其他C原子则以自由C的形式存在于材料中. ...
... 为了最大程度的保留Si-C键, 减少自由C的含量, 可选用含有Si-H基团的有机硅氧烷作为先驱体.有机硅氧烷先驱体中含有Si-H基团, 在减少Si-O键数量的同时, 还会引起 Si-H与Si-Cx y 2 -Si≡键合形式的网络聚合结构.Singh等[48 ] 以甲基二甲氧基硅烷(methyldimethoxysilane, MDMS)和TEOS为双先驱体, 在酸性条件下水解和缩聚后, 得到了多孔SiCO凝胶.基于溶解-再沉淀机制(dissolution-reprecipitation mechanism), 经氨水老化处理, 可实现凝胶孔结构的调节.研究表明, 氨水老化处理后, 凝胶中的(CH3 )HSiO2 结构将转变为(CH3 )SiO3 ; 经高温热解后, 相较于未经氨水老化处理的凝胶来说, 结构中存在更少量的CSiO3 和C2 SiO2 .Si-H键的存在将通过与Si-CH3 反应生成≡Si-CH2 -Si≡键合形式, 进而增加SiCO的含量.因此, 在凝胶中需要保留更多的Si-H键. ...
Synthesis and characterization of silicon oxycarbide glasses.
1
1990
... 采用溶胶-凝胶法合成SiCO是一种最佳的制备方法.为了在这一过程中均匀地引入Si-C键, 常以带有直接与Si原子相连烃基的有机硅氧烷作为先驱体, 即烃基取代硅氧烷先驱体(alkyl-substituted silicon-alkoxide precursors)[46 ] .该先驱体的分子式可表示为Rx 4- x x 可取1~3), 其中, R既可为饱和烃基(如CH3 、C2 H5 ), 也可为不饱和烃基(如C6 H5 ); R’主要为CH3 或C2 H5 [48 ] .在溶胶-凝胶过程中, 与Si直接相连的R并不参与水解, 因而可保留在以Si-O-Si为主体构成的网络结构中.R中含有的碳链长短和数量, 将决定SiCO中Si-C键的数量, 因此, 先驱体的分子组成是影响SiCO最终构成的最直接因素.有研究表明[49 ] , 凝胶中C含量会随着R基团中碳链长度的增加而呈比例增加.但是, 热解后保留的C含量与先驱体或凝胶中存在的碳链长度并无明确比例关系.随着R基团中C含量的降低, C-Si-O键所占比例将增加.热解后, 只有直接与Si原子相连的C原子才可以SiCO的形式保留在材料中, 而其他C原子则以自由C的形式存在于材料中. ...
Dimethyldiethoxysilane/tetraethoxysilane copolymers: precursors for the silicon-carbon-oxygen system.
3
1989
... 通常, 烃基取代硅氧烷先驱体在水解缩聚过程中, 由于不可水解R基团的存在, 往往对硅烷醇的缩聚造成阻碍, 从而使其难以凝胶.因此, 在溶胶-凝胶阶段常借助易于水解缩聚的四烷氧基硅烷(如TMOS、TEOS)构成交联网络结构.Babonneau等[50 ] 采用二甲基二乙氧基硅烷(dimethyldiethoxysilane, DEDMS)和TEOS作为双先驱体, 经水解缩聚后形成了Si-C-O凝胶体系.29 Si核磁共振(nuclear magnetic resonance, NMR)证明这两种双先驱体之间发生了共聚合化作用, 并存在两类凝胶单元, 即源于DEDMS构成的(CH3 )2 Si(O0.5 )2 和源于TEOS构成的Si(O0.5 )4 .Liu等[42 ] 采用苯基三甲氧基硅烷(phenyltrimethoxysilane, PhTMS)和TMOS作为双先驱体制备的凝胶体系由O3/2 -Si-C6 H5 和1/2 O-Si-O1/2 凝胶单元构成.随着PhTMS含量的增加, 孔大小、孔体积和比表面积均下降, 经1000℃热解后, 获得的SiCO仍旧保持 581 m2 /g的高比表面积.Feng等[51 ] 以TEOS和聚二甲基硅氧烷(polydimethylsiloxane, PDMS)为双先驱体, 经溶胶-凝胶、超临界干燥和1200℃热解后得到块状SiCO气凝胶单体.该材料呈非晶态, 其网络结构可在1100℃下保持稳定, 当温度达到1200℃, 非晶态中逐渐开始出现晶相, 而此时仅有1.65%的质量损失, 可见该材料具有很高的热稳定性.另外, 该材料的密度为0.3 g/cm3 , 热导率仅为0.027 W/(m·K), 可作为一种高温隔热材料使用. ...
... 高温热解后, Si、O和C三种元素间的化学构成和分布是SiCO研究中的重要方面.由于热解过程常伴随着剧烈的化学反应和材料收缩, 获得的SiCO致密性往往较高.在热解过程中, 温度高于300℃时, Si-C键开始断裂, 并逐渐向Si-O键转变.SiO2 网络结构中存在的Si-C键一直可保持到900℃[50 ] .经800℃~1000℃高温热解后, SiCO主要由具有自由网络形式的Si-O键和Si-C键构成, 其形式为不同比例的SiC4 、SiOC3 、SiO2 C2 、SiO3 C和SiO4 , 并且含有一定量的自由C.这一复合成分常可表示为: SiCx 2(1- x ) +y Cfree [60 -61 ] .当热解温度高于1000℃~1200℃后, SiCO将发生相变, 形成SiC、C(turbostratic)和SiO2 (cristobalite)三相的平衡.在这一相变过程中, 非晶SiC和C出现在非晶SiO2 或SiCO基体中[60 ] .热解温度高于1300℃时, SiO2 在碳热还原作用下再次生成Si-C键, 达到1500℃后, 可获得晶态SiC[50 ] .Bréquel等[61 ] 为了研究不同化学计量比下SiC和SiO2 复合材料的高温晶化行为, 通过改变双先驱体MDES和TrEOS两者间的相对含量, 设计了三种凝胶配比, 即满足SiCO化学计量的配比、富碳配比和富硅配比.研究表明, 高C含量下, SiCO易于转变为β-SiC晶体; 低C含量下, 易于获得大尺寸晶粒.C的存在将阻碍SiC晶核与SiCO相之间的反应, 因而C富集区域内SiCO转变为SiC的速率变缓.除此以外, SiCO中SiO2 相的高温稳定性也能得以提高[61 ] .造成这一现象的原因可能是SiO2 结构中残余Si-C键可以阻碍SiO2 结构的晶化重组, 并且由于SiO2 团簇的尺寸太小, 难以形成连续的SiO2 基体.通常, 先驱体中与Si直接相连的烃基类型决定了自由C的含量[62 ] .饱和烃基基团通常形成适量的自由C, 不饱和烃基基团易于形成更高含量的自由C.当C含量足够高, 由于碳热还原反应的作用, 将使SiO2 转变为SiC, 同时释放CO. ...
... [50 ].Bréquel等[61 ] 为了研究不同化学计量比下SiC和SiO2 复合材料的高温晶化行为, 通过改变双先驱体MDES和TrEOS两者间的相对含量, 设计了三种凝胶配比, 即满足SiCO化学计量的配比、富碳配比和富硅配比.研究表明, 高C含量下, SiCO易于转变为β-SiC晶体; 低C含量下, 易于获得大尺寸晶粒.C的存在将阻碍SiC晶核与SiCO相之间的反应, 因而C富集区域内SiCO转变为SiC的速率变缓.除此以外, SiCO中SiO2 相的高温稳定性也能得以提高[61 ] .造成这一现象的原因可能是SiO2 结构中残余Si-C键可以阻碍SiO2 结构的晶化重组, 并且由于SiO2 团簇的尺寸太小, 难以形成连续的SiO2 基体.通常, 先驱体中与Si直接相连的烃基类型决定了自由C的含量[62 ] .饱和烃基基团通常形成适量的自由C, 不饱和烃基基团易于形成更高含量的自由C.当C含量足够高, 由于碳热还原反应的作用, 将使SiO2 转变为SiC, 同时释放CO. ...
Synthesis, structure, and properties of silicon oxycarbide aerogels derived from tetraethylortosilicate/polydimethylsiloxane.
2
2015
... 通常, 烃基取代硅氧烷先驱体在水解缩聚过程中, 由于不可水解R基团的存在, 往往对硅烷醇的缩聚造成阻碍, 从而使其难以凝胶.因此, 在溶胶-凝胶阶段常借助易于水解缩聚的四烷氧基硅烷(如TMOS、TEOS)构成交联网络结构.Babonneau等[50 ] 采用二甲基二乙氧基硅烷(dimethyldiethoxysilane, DEDMS)和TEOS作为双先驱体, 经水解缩聚后形成了Si-C-O凝胶体系.29 Si核磁共振(nuclear magnetic resonance, NMR)证明这两种双先驱体之间发生了共聚合化作用, 并存在两类凝胶单元, 即源于DEDMS构成的(CH3 )2 Si(O0.5 )2 和源于TEOS构成的Si(O0.5 )4 .Liu等[42 ] 采用苯基三甲氧基硅烷(phenyltrimethoxysilane, PhTMS)和TMOS作为双先驱体制备的凝胶体系由O3/2 -Si-C6 H5 和1/2 O-Si-O1/2 凝胶单元构成.随着PhTMS含量的增加, 孔大小、孔体积和比表面积均下降, 经1000℃热解后, 获得的SiCO仍旧保持 581 m2 /g的高比表面积.Feng等[51 ] 以TEOS和聚二甲基硅氧烷(polydimethylsiloxane, PDMS)为双先驱体, 经溶胶-凝胶、超临界干燥和1200℃热解后得到块状SiCO气凝胶单体.该材料呈非晶态, 其网络结构可在1100℃下保持稳定, 当温度达到1200℃, 非晶态中逐渐开始出现晶相, 而此时仅有1.65%的质量损失, 可见该材料具有很高的热稳定性.另外, 该材料的密度为0.3 g/cm3 , 热导率仅为0.027 W/(m·K), 可作为一种高温隔热材料使用. ...
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
Thermal stability of periodic mesoporous SiCO glasses.
3
2005
... 除此以外, 为了克服硅氧烷聚合物先驱体热解时的高致密倾向, 多孔SiCO气凝胶型材料逐渐成为研究中的热点.该研究目前主要通过选用[52 -58 ] 或合成[59 ] 不同类型的硅氧烷先驱体, 经溶胶-凝胶、干燥以及高温裂解后, 制备具有多孔结构特征的SiCO.表1 总结了以不同硅氧烷先驱体合成SiCO的孔结构参数, 从中可以看到, SiCO的比表面积和孔体积往往随着裂解温度的升高而降低, 密度则随着裂解温度的升高而增大.这与高温裂解造成材料收缩和致密化有关.另外, 有研究表明, 在400~600℃, 由于热分配反应(thermal redistribution reactions)存在气体释放过程, 往往形成新的临时微孔, 进而提高比表面积和孔体积[53 ,58 ] .温度继续升高后, 新的交联反应引起孔隙收缩, 造成孔结构参数降低[53 ] . ...
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
... [
52 ]
as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 - Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
Surface changes during pyrolytic conversion of hybrid materials to oxycarbide glasses.
3
2009
... 除此以外, 为了克服硅氧烷聚合物先驱体热解时的高致密倾向, 多孔SiCO气凝胶型材料逐渐成为研究中的热点.该研究目前主要通过选用[52 -58 ] 或合成[59 ] 不同类型的硅氧烷先驱体, 经溶胶-凝胶、干燥以及高温裂解后, 制备具有多孔结构特征的SiCO.表1 总结了以不同硅氧烷先驱体合成SiCO的孔结构参数, 从中可以看到, SiCO的比表面积和孔体积往往随着裂解温度的升高而降低, 密度则随着裂解温度的升高而增大.这与高温裂解造成材料收缩和致密化有关.另外, 有研究表明, 在400~600℃, 由于热分配反应(thermal redistribution reactions)存在气体释放过程, 往往形成新的临时微孔, 进而提高比表面积和孔体积[53 ,58 ] .温度继续升高后, 新的交联反应引起孔隙收缩, 造成孔结构参数降低[53 ] . ...
... [53 ]. ...
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
Gels dried under supercritical and ambient conditions: a comparative study and their subsequent conversion to silica-carbon composite aerogels.
1
2013
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
High rate capability of SiOC ceramic aerogels with tailored porosity as anode materials for Li-ion batteries.
1
2015
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
Porous silicon oxycarbide glasses from hybrid ambigels.
1
2011
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
Surface and structural modification of nanostructured mesoporous silicon oxycarbide glasses obtained from preceramic hybrids aged in NH4 OH.
1
2013
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
Influence of the microstructure on the high temperature behaviour of gel-derived SiOC glasses.
3
2001
... 除此以外, 为了克服硅氧烷聚合物先驱体热解时的高致密倾向, 多孔SiCO气凝胶型材料逐渐成为研究中的热点.该研究目前主要通过选用[52 -58 ] 或合成[59 ] 不同类型的硅氧烷先驱体, 经溶胶-凝胶、干燥以及高温裂解后, 制备具有多孔结构特征的SiCO.表1 总结了以不同硅氧烷先驱体合成SiCO的孔结构参数, 从中可以看到, SiCO的比表面积和孔体积往往随着裂解温度的升高而降低, 密度则随着裂解温度的升高而增大.这与高温裂解造成材料收缩和致密化有关.另外, 有研究表明, 在400~600℃, 由于热分配反应(thermal redistribution reactions)存在气体释放过程, 往往形成新的临时微孔, 进而提高比表面积和孔体积[53 ,58 ] .温度继续升高后, 新的交联反应引起孔隙收缩, 造成孔结构参数降低[53 ] . ...
... ,58 ].温度继续升高后, 新的交联反应引起孔隙收缩, 造成孔结构参数降低[53 ] . ...
... Porous parameters of SiCO prepared by different siloxane precursors
Precursors Temperature/ Density/-3 ) Ratio of porosity/ specific surface area/(m2 •g-1 ) Pore volume/3 •g-1 ) Average pore size/nm PhTMS+TMOS[42 ] as-prepared 0.48 - 987 - 2.8 TEOS+PDMS[51 ] 1200 0.30 - 198.04 0.684 5.6 MDMS+TEOS[46 ] as-prepared - - 425.5 1.87 17.59 BTEE[52 ] as-prepared - - 1022 0.53 - BTME[52 ] as-prepared - - 867 0.74 - TEOS+TBOT+PDMS[53 ] as-prepared - - 1.1 1.7 - BTEBP[13 ] 300 0.264 83 1190 0.916 - MTMS+GPYMS[54 ] as-prepared 0.31 78 464 1.24 11 PHMS[55 ] as-prepared - - 227 1.37 52 MTES[56 ] as-prepared - - 727 1.47 8.0 PDMS+TrEOS[57 ] as-prepared - 59-69 405-583 - 3.2-5.0 MDES+TrEOS[58 ] as-prepared - 88±2 0.45±0.02 -
Note: BTEE: 1,2-Bis(triethoxysilyl)ethane; BTME: 1,2-bis(trimethoxysilyl)ethane; TBOT: tetrabutyl orthotitanate; BTEBP: 4,4’-bis (triethoxysilyl)-1,1’-biphenyl; MTMS: methyltrimethoxysilane; GPTMS: 3-(2,3-epoxypropoxy) propyltrimethoxysilane; PHMS: polyhydridomethyl siloxane; MDES: methyldiethoxysilane; TrEOS: triethoxysilane ...
Sol- Gel Processing of a glycolated cyclic organosilane and its pyrolysis to silicon oxycarbide monoliths with multiscale porosity and large surface areas.
1
2010
... 除此以外, 为了克服硅氧烷聚合物先驱体热解时的高致密倾向, 多孔SiCO气凝胶型材料逐渐成为研究中的热点.该研究目前主要通过选用[52 -58 ] 或合成[59 ] 不同类型的硅氧烷先驱体, 经溶胶-凝胶、干燥以及高温裂解后, 制备具有多孔结构特征的SiCO.表1 总结了以不同硅氧烷先驱体合成SiCO的孔结构参数, 从中可以看到, SiCO的比表面积和孔体积往往随着裂解温度的升高而降低, 密度则随着裂解温度的升高而增大.这与高温裂解造成材料收缩和致密化有关.另外, 有研究表明, 在400~600℃, 由于热分配反应(thermal redistribution reactions)存在气体释放过程, 往往形成新的临时微孔, 进而提高比表面积和孔体积[53 ,58 ] .温度继续升高后, 新的交联反应引起孔隙收缩, 造成孔结构参数降低[53 ] . ...
Chemical durability of silicon oxycarbide glasses.
3
2002
... 高温热解后, Si、O和C三种元素间的化学构成和分布是SiCO研究中的重要方面.由于热解过程常伴随着剧烈的化学反应和材料收缩, 获得的SiCO致密性往往较高.在热解过程中, 温度高于300℃时, Si-C键开始断裂, 并逐渐向Si-O键转变.SiO2 网络结构中存在的Si-C键一直可保持到900℃[50 ] .经800℃~1000℃高温热解后, SiCO主要由具有自由网络形式的Si-O键和Si-C键构成, 其形式为不同比例的SiC4 、SiOC3 、SiO2 C2 、SiO3 C和SiO4 , 并且含有一定量的自由C.这一复合成分常可表示为: SiCx 2(1- x ) +y Cfree [60 -61 ] .当热解温度高于1000℃~1200℃后, SiCO将发生相变, 形成SiC、C(turbostratic)和SiO2 (cristobalite)三相的平衡.在这一相变过程中, 非晶SiC和C出现在非晶SiO2 或SiCO基体中[60 ] .热解温度高于1300℃时, SiO2 在碳热还原作用下再次生成Si-C键, 达到1500℃后, 可获得晶态SiC[50 ] .Bréquel等[61 ] 为了研究不同化学计量比下SiC和SiO2 复合材料的高温晶化行为, 通过改变双先驱体MDES和TrEOS两者间的相对含量, 设计了三种凝胶配比, 即满足SiCO化学计量的配比、富碳配比和富硅配比.研究表明, 高C含量下, SiCO易于转变为β-SiC晶体; 低C含量下, 易于获得大尺寸晶粒.C的存在将阻碍SiC晶核与SiCO相之间的反应, 因而C富集区域内SiCO转变为SiC的速率变缓.除此以外, SiCO中SiO2 相的高温稳定性也能得以提高[61 ] .造成这一现象的原因可能是SiO2 结构中残余Si-C键可以阻碍SiO2 结构的晶化重组, 并且由于SiO2 团簇的尺寸太小, 难以形成连续的SiO2 基体.通常, 先驱体中与Si直接相连的烃基类型决定了自由C的含量[62 ] .饱和烃基基团通常形成适量的自由C, 不饱和烃基基团易于形成更高含量的自由C.当C含量足够高, 由于碳热还原反应的作用, 将使SiO2 转变为SiC, 同时释放CO. ...
... [60 ].热解温度高于1300℃时, SiO2 在碳热还原作用下再次生成Si-C键, 达到1500℃后, 可获得晶态SiC[50 ] .Bréquel等[61 ] 为了研究不同化学计量比下SiC和SiO2 复合材料的高温晶化行为, 通过改变双先驱体MDES和TrEOS两者间的相对含量, 设计了三种凝胶配比, 即满足SiCO化学计量的配比、富碳配比和富硅配比.研究表明, 高C含量下, SiCO易于转变为β-SiC晶体; 低C含量下, 易于获得大尺寸晶粒.C的存在将阻碍SiC晶核与SiCO相之间的反应, 因而C富集区域内SiCO转变为SiC的速率变缓.除此以外, SiCO中SiO2 相的高温稳定性也能得以提高[61 ] .造成这一现象的原因可能是SiO2 结构中残余Si-C键可以阻碍SiO2 结构的晶化重组, 并且由于SiO2 团簇的尺寸太小, 难以形成连续的SiO2 基体.通常, 先驱体中与Si直接相连的烃基类型决定了自由C的含量[62 ] .饱和烃基基团通常形成适量的自由C, 不饱和烃基基团易于形成更高含量的自由C.当C含量足够高, 由于碳热还原反应的作用, 将使SiO2 转变为SiC, 同时释放CO. ...
... 经热解法制备的SiCO比SiO2 具有更高的力学、热学和化学稳定性[46 ] .在SiCO中, C以自由碳或以与Si连接形成SiC4- x x 4 基团的形式存在.相比于硅氧间只能形成二氧连接, 硅碳间最多的四碳连接形式则可形成更为坚固的刚性骨架, 从而提高SiCO的模量、硬度、粘度、密度和玻璃转化温度.当体系中的自由C含量逐渐升高时, SiCO的弹性模量、弯曲强度、维氏硬度都会出现明显增加[63 -65 ] .表2 比较了SiCO和玻璃态SiO2 之间的物理特性[64 ] .在化学稳定性方面, SiCO经历如碱性条件或HF的腐蚀后, 由于其中的Si-C键可经受更高的亲核攻击(nucleophilic attack), 具有更高的无序和交联度, 并且与Si原子相连的C和自由C均可对腐蚀反应起阻碍作用, 因而比SiO2 表现出更好的化学稳定性[60 ] .针对多孔SiCO的传热问题, Qiu等[66 ] 建立了一种描述其热传递的立方阵列交叉纳米球模型(cubic array of an intersecting nano-spheres model), 并发展了预测有效热导率的理论公式.结果表明, 多孔SiCO室温下的有效热导率在0.041~0.078 W/(m·K)之间, 该数值主要与SiCO固相体积分数以及相邻颗粒间接触长度(a )与颗粒直径(d )的比值a /d 有关.图5 给出了该研究中建立的多孔SiCO陶瓷的几何结构和热传递分析模型[66 ] . ...
Systematic structural characterization of the high-temperature behavior of nearly stoichiometric silicon oxycarbide glasses.
3
2004
... 高温热解后, Si、O和C三种元素间的化学构成和分布是SiCO研究中的重要方面.由于热解过程常伴随着剧烈的化学反应和材料收缩, 获得的SiCO致密性往往较高.在热解过程中, 温度高于300℃时, Si-C键开始断裂, 并逐渐向Si-O键转变.SiO2 网络结构中存在的Si-C键一直可保持到900℃[50 ] .经800℃~1000℃高温热解后, SiCO主要由具有自由网络形式的Si-O键和Si-C键构成, 其形式为不同比例的SiC4 、SiOC3 、SiO2 C2 、SiO3 C和SiO4 , 并且含有一定量的自由C.这一复合成分常可表示为: SiCx 2(1- x ) +y Cfree [60 -61 ] .当热解温度高于1000℃~1200℃后, SiCO将发生相变, 形成SiC、C(turbostratic)和SiO2 (cristobalite)三相的平衡.在这一相变过程中, 非晶SiC和C出现在非晶SiO2 或SiCO基体中[60 ] .热解温度高于1300℃时, SiO2 在碳热还原作用下再次生成Si-C键, 达到1500℃后, 可获得晶态SiC[50 ] .Bréquel等[61 ] 为了研究不同化学计量比下SiC和SiO2 复合材料的高温晶化行为, 通过改变双先驱体MDES和TrEOS两者间的相对含量, 设计了三种凝胶配比, 即满足SiCO化学计量的配比、富碳配比和富硅配比.研究表明, 高C含量下, SiCO易于转变为β-SiC晶体; 低C含量下, 易于获得大尺寸晶粒.C的存在将阻碍SiC晶核与SiCO相之间的反应, 因而C富集区域内SiCO转变为SiC的速率变缓.除此以外, SiCO中SiO2 相的高温稳定性也能得以提高[61 ] .造成这一现象的原因可能是SiO2 结构中残余Si-C键可以阻碍SiO2 结构的晶化重组, 并且由于SiO2 团簇的尺寸太小, 难以形成连续的SiO2 基体.通常, 先驱体中与Si直接相连的烃基类型决定了自由C的含量[62 ] .饱和烃基基团通常形成适量的自由C, 不饱和烃基基团易于形成更高含量的自由C.当C含量足够高, 由于碳热还原反应的作用, 将使SiO2 转变为SiC, 同时释放CO. ...
... [61 ]为了研究不同化学计量比下SiC和SiO2 复合材料的高温晶化行为, 通过改变双先驱体MDES和TrEOS两者间的相对含量, 设计了三种凝胶配比, 即满足SiCO化学计量的配比、富碳配比和富硅配比.研究表明, 高C含量下, SiCO易于转变为β-SiC晶体; 低C含量下, 易于获得大尺寸晶粒.C的存在将阻碍SiC晶核与SiCO相之间的反应, 因而C富集区域内SiCO转变为SiC的速率变缓.除此以外, SiCO中SiO2 相的高温稳定性也能得以提高[61 ] .造成这一现象的原因可能是SiO2 结构中残余Si-C键可以阻碍SiO2 结构的晶化重组, 并且由于SiO2 团簇的尺寸太小, 难以形成连续的SiO2 基体.通常, 先驱体中与Si直接相连的烃基类型决定了自由C的含量[62 ] .饱和烃基基团通常形成适量的自由C, 不饱和烃基基团易于形成更高含量的自由C.当C含量足够高, 由于碳热还原反应的作用, 将使SiO2 转变为SiC, 同时释放CO. ...
... [61 ].造成这一现象的原因可能是SiO2 结构中残余Si-C键可以阻碍SiO2 结构的晶化重组, 并且由于SiO2 团簇的尺寸太小, 难以形成连续的SiO2 基体.通常, 先驱体中与Si直接相连的烃基类型决定了自由C的含量[62 ] .饱和烃基基团通常形成适量的自由C, 不饱和烃基基团易于形成更高含量的自由C.当C含量足够高, 由于碳热还原反应的作用, 将使SiO2 转变为SiC, 同时释放CO. ...
HOURLIER‐BAHLOUL D, et al. Silicon oxycarbide glasses: Part 1-Thermochemical stability.
1
2006
... 高温热解后, Si、O和C三种元素间的化学构成和分布是SiCO研究中的重要方面.由于热解过程常伴随着剧烈的化学反应和材料收缩, 获得的SiCO致密性往往较高.在热解过程中, 温度高于300℃时, Si-C键开始断裂, 并逐渐向Si-O键转变.SiO2 网络结构中存在的Si-C键一直可保持到900℃[50 ] .经800℃~1000℃高温热解后, SiCO主要由具有自由网络形式的Si-O键和Si-C键构成, 其形式为不同比例的SiC4 、SiOC3 、SiO2 C2 、SiO3 C和SiO4 , 并且含有一定量的自由C.这一复合成分常可表示为: SiCx 2(1- x ) +y Cfree [60 -61 ] .当热解温度高于1000℃~1200℃后, SiCO将发生相变, 形成SiC、C(turbostratic)和SiO2 (cristobalite)三相的平衡.在这一相变过程中, 非晶SiC和C出现在非晶SiO2 或SiCO基体中[60 ] .热解温度高于1300℃时, SiO2 在碳热还原作用下再次生成Si-C键, 达到1500℃后, 可获得晶态SiC[50 ] .Bréquel等[61 ] 为了研究不同化学计量比下SiC和SiO2 复合材料的高温晶化行为, 通过改变双先驱体MDES和TrEOS两者间的相对含量, 设计了三种凝胶配比, 即满足SiCO化学计量的配比、富碳配比和富硅配比.研究表明, 高C含量下, SiCO易于转变为β-SiC晶体; 低C含量下, 易于获得大尺寸晶粒.C的存在将阻碍SiC晶核与SiCO相之间的反应, 因而C富集区域内SiCO转变为SiC的速率变缓.除此以外, SiCO中SiO2 相的高温稳定性也能得以提高[61 ] .造成这一现象的原因可能是SiO2 结构中残余Si-C键可以阻碍SiO2 结构的晶化重组, 并且由于SiO2 团簇的尺寸太小, 难以形成连续的SiO2 基体.通常, 先驱体中与Si直接相连的烃基类型决定了自由C的含量[62 ] .饱和烃基基团通常形成适量的自由C, 不饱和烃基基团易于形成更高含量的自由C.当C含量足够高, 由于碳热还原反应的作用, 将使SiO2 转变为SiC, 同时释放CO. ...
D'ANDREA G. Mechanical characterization of Sol-Gel-derived silicon oxycarbide glasses.
1
1996
... 经热解法制备的SiCO比SiO2 具有更高的力学、热学和化学稳定性[46 ] .在SiCO中, C以自由碳或以与Si连接形成SiC4- x x 4 基团的形式存在.相比于硅氧间只能形成二氧连接, 硅碳间最多的四碳连接形式则可形成更为坚固的刚性骨架, 从而提高SiCO的模量、硬度、粘度、密度和玻璃转化温度.当体系中的自由C含量逐渐升高时, SiCO的弹性模量、弯曲强度、维氏硬度都会出现明显增加[63 -65 ] .表2 比较了SiCO和玻璃态SiO2 之间的物理特性[64 ] .在化学稳定性方面, SiCO经历如碱性条件或HF的腐蚀后, 由于其中的Si-C键可经受更高的亲核攻击(nucleophilic attack), 具有更高的无序和交联度, 并且与Si原子相连的C和自由C均可对腐蚀反应起阻碍作用, 因而比SiO2 表现出更好的化学稳定性[60 ] .针对多孔SiCO的传热问题, Qiu等[66 ] 建立了一种描述其热传递的立方阵列交叉纳米球模型(cubic array of an intersecting nano-spheres model), 并发展了预测有效热导率的理论公式.结果表明, 多孔SiCO室温下的有效热导率在0.041~0.078 W/(m·K)之间, 该数值主要与SiCO固相体积分数以及相邻颗粒间接触长度(a )与颗粒直径(d )的比值a /d 有关.图5 给出了该研究中建立的多孔SiCO陶瓷的几何结构和热传递分析模型[66 ] . ...
Silicon oxycarbide glasses: Part II. Structure and properties.
1
1991
... 经热解法制备的SiCO比SiO2 具有更高的力学、热学和化学稳定性[46 ] .在SiCO中, C以自由碳或以与Si连接形成SiC4- x x 4 基团的形式存在.相比于硅氧间只能形成二氧连接, 硅碳间最多的四碳连接形式则可形成更为坚固的刚性骨架, 从而提高SiCO的模量、硬度、粘度、密度和玻璃转化温度.当体系中的自由C含量逐渐升高时, SiCO的弹性模量、弯曲强度、维氏硬度都会出现明显增加[63 -65 ] .表2 比较了SiCO和玻璃态SiO2 之间的物理特性[64 ] .在化学稳定性方面, SiCO经历如碱性条件或HF的腐蚀后, 由于其中的Si-C键可经受更高的亲核攻击(nucleophilic attack), 具有更高的无序和交联度, 并且与Si原子相连的C和自由C均可对腐蚀反应起阻碍作用, 因而比SiO2 表现出更好的化学稳定性[60 ] .针对多孔SiCO的传热问题, Qiu等[66 ] 建立了一种描述其热传递的立方阵列交叉纳米球模型(cubic array of an intersecting nano-spheres model), 并发展了预测有效热导率的理论公式.结果表明, 多孔SiCO室温下的有效热导率在0.041~0.078 W/(m·K)之间, 该数值主要与SiCO固相体积分数以及相邻颗粒间接触长度(a )与颗粒直径(d )的比值a /d 有关.图5 给出了该研究中建立的多孔SiCO陶瓷的几何结构和热传递分析模型[66 ] . ...
Mechanical characterization of a polysiloxane-derived SiOC glass.
1
2007
... 经热解法制备的SiCO比SiO2 具有更高的力学、热学和化学稳定性[46 ] .在SiCO中, C以自由碳或以与Si连接形成SiC4- x x 4 基团的形式存在.相比于硅氧间只能形成二氧连接, 硅碳间最多的四碳连接形式则可形成更为坚固的刚性骨架, 从而提高SiCO的模量、硬度、粘度、密度和玻璃转化温度.当体系中的自由C含量逐渐升高时, SiCO的弹性模量、弯曲强度、维氏硬度都会出现明显增加[63 -65 ] .表2 比较了SiCO和玻璃态SiO2 之间的物理特性[64 ] .在化学稳定性方面, SiCO经历如碱性条件或HF的腐蚀后, 由于其中的Si-C键可经受更高的亲核攻击(nucleophilic attack), 具有更高的无序和交联度, 并且与Si原子相连的C和自由C均可对腐蚀反应起阻碍作用, 因而比SiO2 表现出更好的化学稳定性[60 ] .针对多孔SiCO的传热问题, Qiu等[66 ] 建立了一种描述其热传递的立方阵列交叉纳米球模型(cubic array of an intersecting nano-spheres model), 并发展了预测有效热导率的理论公式.结果表明, 多孔SiCO室温下的有效热导率在0.041~0.078 W/(m·K)之间, 该数值主要与SiCO固相体积分数以及相邻颗粒间接触长度(a )与颗粒直径(d )的比值a /d 有关.图5 给出了该研究中建立的多孔SiCO陶瓷的几何结构和热传递分析模型[66 ] . ...
Thermal-conductivity studies of macro-porous polymer-derived SiOC ceramics.
4
2014
... 经热解法制备的SiCO比SiO2 具有更高的力学、热学和化学稳定性[46 ] .在SiCO中, C以自由碳或以与Si连接形成SiC4- x x 4 基团的形式存在.相比于硅氧间只能形成二氧连接, 硅碳间最多的四碳连接形式则可形成更为坚固的刚性骨架, 从而提高SiCO的模量、硬度、粘度、密度和玻璃转化温度.当体系中的自由C含量逐渐升高时, SiCO的弹性模量、弯曲强度、维氏硬度都会出现明显增加[63 -65 ] .表2 比较了SiCO和玻璃态SiO2 之间的物理特性[64 ] .在化学稳定性方面, SiCO经历如碱性条件或HF的腐蚀后, 由于其中的Si-C键可经受更高的亲核攻击(nucleophilic attack), 具有更高的无序和交联度, 并且与Si原子相连的C和自由C均可对腐蚀反应起阻碍作用, 因而比SiO2 表现出更好的化学稳定性[60 ] .针对多孔SiCO的传热问题, Qiu等[66 ] 建立了一种描述其热传递的立方阵列交叉纳米球模型(cubic array of an intersecting nano-spheres model), 并发展了预测有效热导率的理论公式.结果表明, 多孔SiCO室温下的有效热导率在0.041~0.078 W/(m·K)之间, 该数值主要与SiCO固相体积分数以及相邻颗粒间接触长度(a )与颗粒直径(d )的比值a /d 有关.图5 给出了该研究中建立的多孔SiCO陶瓷的几何结构和热传递分析模型[66 ] . ...
... [66 ]. ...
... 多孔SiCO陶瓷的几何结构和热传递分析模型[66 ] ...
... Geometric structure and heat transfer analysis of macro-porous SiCO ceramics[66 ] ...
 , 李亚
, 李亚
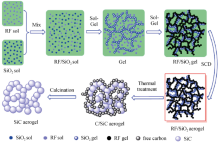
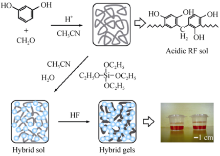
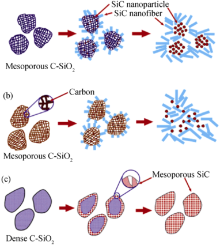
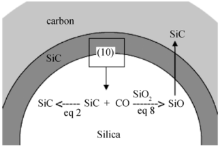
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}