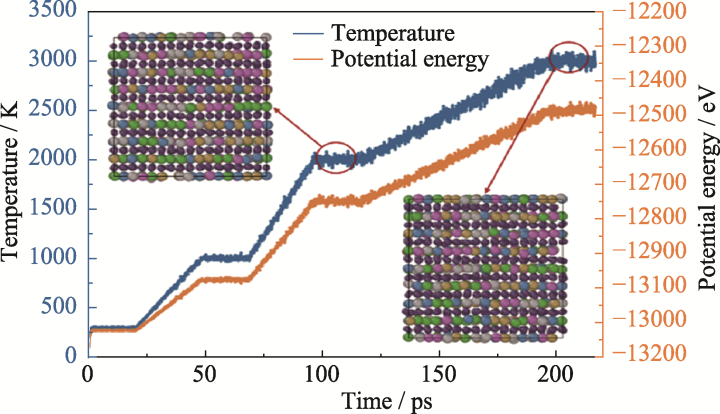
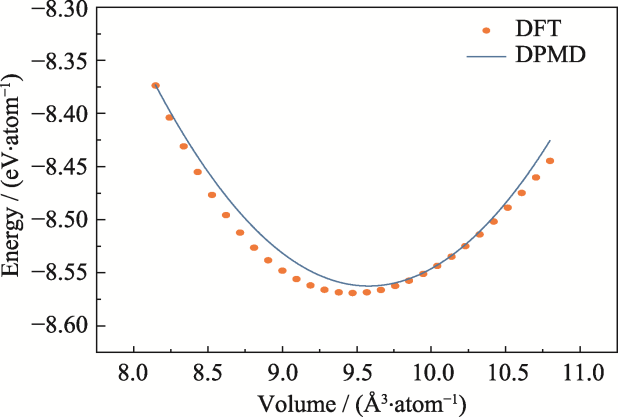
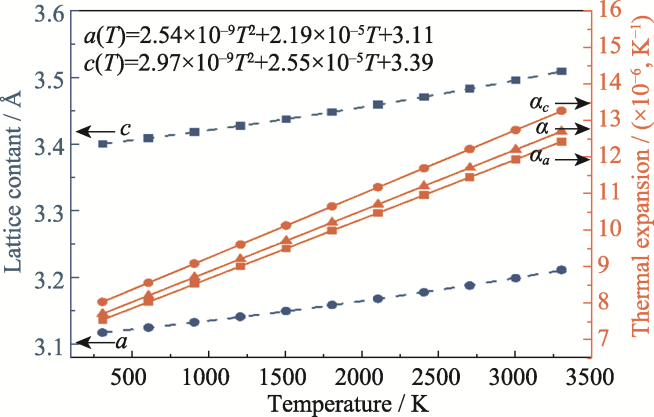
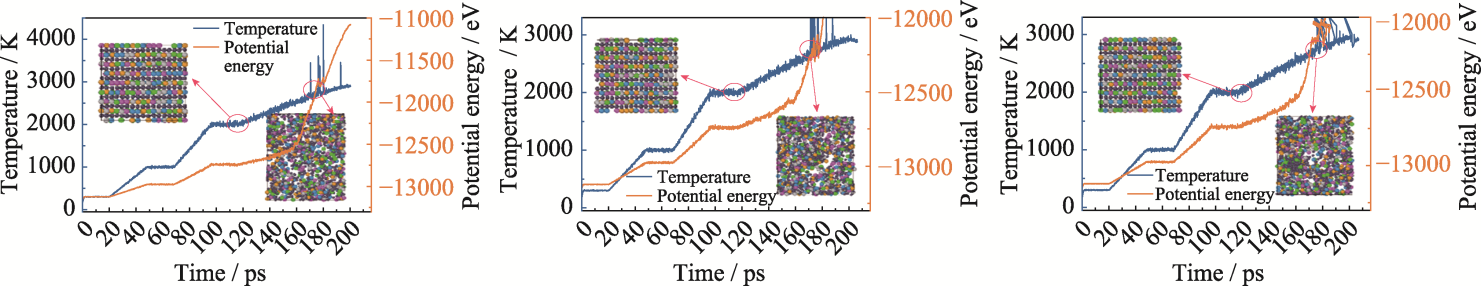
| [1] |
贾成科, 张鑫, 任先京, 等. 硼化物陶瓷基涂层制备技术的研究进展. 热喷涂技术, 2012, 4(1): 5.
|
| [2] |
MONTEVERDE F, GUICCIARDI S, BELLOSI A. Advances in microstructure and mechanical properties of zirconium diboride based ceramics. Materials Science and Engineering: A, 2003, 346(1): 310.
DOI
URL
|
| [3] |
WU W W, ZHANG G J, KAN Y M, et al. Synthesis and microstructural features of ZrB2-SiC-based composites by reactive spark plasma sintering and reactive hot pressing. Scripta Materialia, 2007, 57(4): 317.
DOI
URL
|
| [4] |
ZOU J, ZHANG G J, KAN Y M, et al. Pressureless densification of Zrb2-SiC composites with vanadium carbide. Scripta Materialia, 2008, 59(3):309.
DOI
URL
|
| [5] |
YEH J W, CHEN S K, LIN S J, et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Advanced Engineering Materials, 2004, 6(5): 299.
DOI
URL
|
| [6] |
MIRACLE D B, SENKOV O N. A critical review of high entropy alloys and related concepts. Acta Materialia, 2017, 122: 448.
DOI
URL
|
| [7] |
GEORGE E P, RAABE D, RITCHIE R O. High-entropy alloys. Nature Reviews Materials, 2019, 4(8): 515.
DOI
|
| [8] |
WANG Y P, GAN G Y, WANG W, et al. First-principles study of mechanical and thermal properties of high-entropy diborides. Physica Status Solidi (B), 2018, 255(10): 1800011.
DOI
URL
|
| [9] |
ZHANG Y, CHEN G, SHI X, et al. Atomic simulations and experimental of the effect of transition metal on the high- temperature oxidation behavior of ZrB2. Corrosion Science, 2024, 236: 112218.
DOI
URL
|
| [10] |
MENG X Y, ZHANG H X, MEZEI M, et al. Molecular docking: a powerful approach for structure-based drug discovery. Current Computer-Aided Drug Design, 2011, 7(2): 146.
|
| [11] |
ALDER B J, WAINWRIGHT T E. Studies in molecular dynamics. I. General method. Journal of Chemical Physics, 1959, 31(2): 459.
|
| [12] |
BEHLER J, PARRINELLO M. Generalized neural-network representation of potential-energy surfaces. Physical Review Letters, 2007; 98(14): 146401.
|
| [13] |
DAI F Z, SUN Y J, WEN B, et al. Temperature dependent thermal and elastic properties of high entropy (Ti0.2Zr0.2Hf0.2Nb0.2Ta0.2)B2: molecular dynamics simulation by deep learning potential. Journal of Materials Science and Technology, 2021, 72: 8.
DOI
URL
|
| [14] |
ZHANG X, LI B, ZHOU J, et al. Deep-learning enabled atomic insights into the phase transitions and nanodomain topology of lead-free (K,Na)NbO3 ferroelectrics. Science China Materials, 2024, 67(9): 3029.
DOI
|
| [15] |
VAN DE WALLE A, TIWARY P, DE JONG M, et al. Efficient stochastic generation of special quasirandom structures. Calphad, 2013, 42: 13.
DOI
URL
|
| [16] |
THOMPSON A P, AKTULGA H M, BERGER R, et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Computer Physics Communications, 2022, 271: 108171.
DOI
URL
|
| [17] |
GU J, ZOU J, SUN S K, et al. Dense and pure high-entropy metal diboride ceramics sintered from self-synthesized powders via boro/carbothermal reduction approach. Science China Materials, 2019, 62(12): 1898.
DOI
|
| [18] |
LIU J, YANG Q, ZOU J, et al. Strong high-entropy diborides ceramics with oxide impurities at 1800 ℃. Science China Materials, 2023, 66(5): 1892.
|
 ), 张旭1,2, 张小锋3, 李蓓1,2(
), 张旭1,2, 张小锋3, 李蓓1,2(