

本研究使用常用的工业原料合成具有良好电化学活性面积(ECSA)和氧还原反应(ORR)能力的PtCo合金纳米电催化剂; 探究引入Co后, 合成PtCo合金纳米电催化剂所需还原剂(NaBH4)和碱性络合剂(三乙胺)的最佳添加量及高温热处理的最佳温度。
1 实验方法
1.1 典型铂钴合金纳米电催化剂的合成
取1.504 g氯铂酸(H2PtCl6·6H2O, 昆明铂锐金属材料有限公司)水溶液(10wt%)和21.78 mg氯化钴(CoCl2·6H2O, ≥99.8%, 国药集团化学试剂有限公司)加入25 mL无水乙醇(≤0.005%水, 国药集团化学试剂有限公司)中, 超声10 min(60 W, 无锡上佳生物科技有限公司)混合均匀。取100 μL三乙胺((CH2CH3)3N, ≥99.9%, 国药集团化学试剂有限公司)滴入混合液中, 超声至浑浊均匀状态, 得到前驱液。在冰浴条件下, 取57.84 mg碳黑(EC-600JD, 日本狮王株式会社)加入前驱液中, 超声1 h得混合浆料。
混合浆料在磁力搅拌(1000 r/min)条件下, 加入100 mg NaBH4(≥96.0%, 国药集团化学试剂有限公司), 搅拌1 h后, 超纯水洗涤3次, 在80 ℃真空干燥10 h, 得到典型PtCo合金纳米电催化剂, 标记为Pt2.8Co/C。继续对Pt2.8Co/C进行高温热处理: 升温梯度8.3 ℃/min, 在H2浓度为20%的H2/N2混合气条件下, 不同高温处理2 h。将高温热处理的Pt2.8Co/C标记为Pt2.8Co/C-T, 其中T为具体温度。
根据Pt与Co的添加比例, 其它条件不变, 未高温热处理且只改变硼氢化钠添加量所制备的催化剂标记为DN-Pt2.8Co/C; 其它条件不变, 未高温热处理且只改变三乙胺添加量所制备的催化剂标记为DTL-Pt2.8Co/C; 将进行加速稳定性测试后的催化剂样品标记为ADT-M(M为本文中所命名的催化剂)。此外, TKK商用PtCo合金催化剂(TEC36E52, 金属载量52wt%, 其中Pt为47wt%, Co为5wt%)标记为TKK-PtCo/C。本研究中所制备的电催化剂金属载量均与TKK-PtCo/C保持一致。
1.2 催化剂物理表征
使用透射电子显微镜(TEM, TECNAI F20)、能量色散X射线光谱(EDS)、X射线衍射(XRD, Bruker D8 Adavance)对PtCo合金纳米电催化剂的晶型结构、形貌特征和物理特性进行表征。
1.3 电化学性能测试
用Al2O3粉末(0.05 μm)将玻璃碳电极(d=5 mm)表面打磨光滑。取2 mg样品, 加入600 μL超纯水, 超声3 min后, 加入200 μL异丙醇(≥99.9%, 国药集团化学试剂有限公司)。在冰浴条件下, 超声10 min。取50 μL Nafion(5wt%, 美国Cobot公司)滴入混合液中, 超声30 min后取10 μL混合浆料滴至玻璃碳电极表面, 均匀成膜[16]。
干燥成膜后, 采用Gamry公司的Reference 3000电化学工作站进行电化学测试和相关数据记录。采用标准三电极体系测试循环伏安曲线(CV)以计算ECSA, 测试线性伏安曲线(LSV)以计算质量活性(MA), 参比电极为饱和甘汞电极(SCE), 铂丝(d=0.5 mm)为对电极, 工作电极为玻璃碳电极(d= 5 mm, S=0.196 cm2), 电解液为0.1 mol/L的HCLO4水溶液。
在0.1 mol/L的氮气饱和HCLO4溶液中, 进行循环伏安(CV)测试。活化过程: 扫描电位0.05~ 1.2 V(vs RHE), 扫速200 mV/s, 扫描20圈。CV测试过程: 扫描电位0.05~1.2 V(vs RHE), 扫速20 mV/s, 扫描3圈。完成CV测试后, 在0.1 mol/L的氧气饱和HCLO4溶液中, 进行线性伏安曲线(LSV)测试, 扫描电位为0.05~1.05 V(vs RHE), 扫速10 mV/s, 电极旋转速度为1600 r/min。加速稳定性测试(ADT): 0.1 mol/L的氮气饱和HCLO4溶液条件下, 扫描电位为0.6~1.2 V(vs RHE), 扫速100 mV/s, 扫描5000圈。
ECSA和MA的计算方法如下:
其中, Q(C)(mC·cm-2)是氢脱吸附区域的库仑电荷; LPt(mg·cm-2)是玻璃碳电极上的Pt负载质量; Q(AC)= 0.21 mC·cm-2, 表示铂表面单层原子吸附氢的电荷量; ik是由K-L方程得出的动力学电流; mPt是玻璃碳电极上的铂载量。
2 结果与讨论
2.1 物理性能表征
图1为Pt2.8Co/C、Pt2.8Co/C-500和TKK-PtCo/C的XRD图谱。样品都具有Pt和PtCo对应的典型面心立方结构(fcc); Pt2.8Co/C与Pt2.8Co/C-500相应衍射峰的2θ角比标准Pt单晶更大, 与TKK-PtCo/C衍射峰相似且都向右偏移, 证明原子尺寸较小的Co进入了尺寸较大的Pt晶格[17]。经过500 ℃高温热处理后的催化剂, 衍射峰明显增强, 这说明催化剂的结晶度更优; 此外, 衍射峰半高宽度缩小, 也说明催化剂尺寸增大。结合XRD图谱与Bragg公式计算得到, Pt2.8Co/C、Pt2.8Co/C-500和TKK-PtCo/C的Pt(111)晶格间距分别为0.224、0.223和0.221 nm, 与标准单晶Pt(111)的0.226 nm相比均明显减小, 这与XRD衍射峰向右偏移的现象相吻合, 表明Pt和Co之间具有良好的合金化程度, 本工艺成功制备了PtCo合金催化剂。
图1
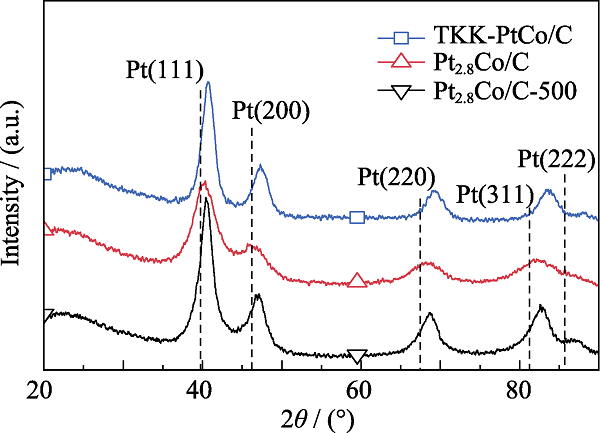
图1
Pt2.8Co/C、Pt2.8Co/C-500和TKK-PtCo/C的XRD图谱
Fig. 1
XRD patterns of Pt2.8Co/C, Pt2.8Co/C-500 and TKK-PtCo/C
图2(a~c)分别为Pt2.8Co/C、Pt2.8Co/C-500、TKK-PtCo/C的TEM照片及其粒径分布图。TEM照片显示的晶格间距与XRD计算结果近似; 其中, Pt2.8Co/C-500的晶格间距较Pt2.8Co/C更小, 说明高温热处理进一步提升了铂钴的合金化程度。三种催化剂的形貌近似球形, Pt2.8Co/C与Pt2.8Co/C-500较为均匀地分布在碳载体上, 且颗粒尺寸差异较小, 但TKK-PtCo/C的颗粒差异较大。通过随机统计100个金属颗粒得到Pt2.8Co/C、Pt2.8Co/C-500的平均颗粒尺寸分别为3.45、3.85 nm, 均小于TKK-PtCo/C (5.75 nm)。纳米金属颗粒更小, 才能更有效地提高Pt的利用率, 提供更多活性位点, 进而提升催化剂的电催化能力[3,18-20]。
图2
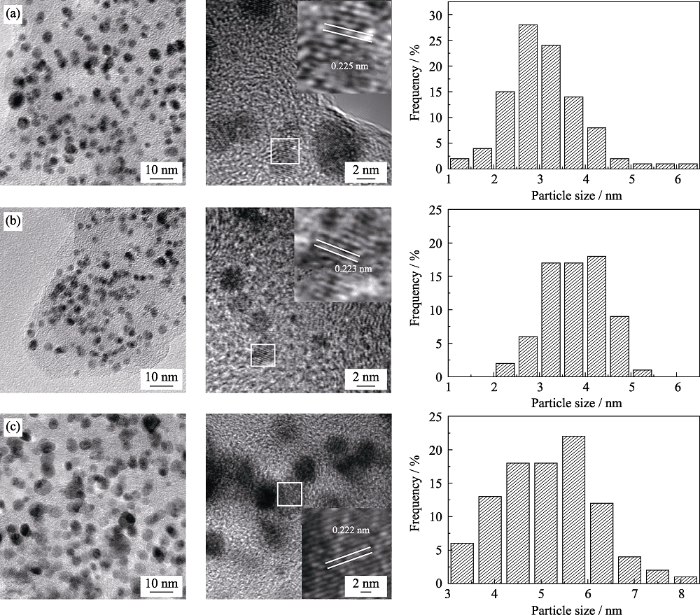
图2
Pt2.8Co/C(a)、Pt2.8Co/C-500(b)和TKK-PtCo/C(c)的TEM照片及粒径分布图
Fig. 2
TEM images and particle size distributions of Pt2.8Co/C(a), Pt2.8Co/C-500(b) and TKK-PtCo/C(c)
图3

图3
Pt2.8Co/C-500的EDS图谱
Fig. 3
EDS mappings of Pt2.8Co/C-500
(a) TEM image; (b) PtCo alloy; (c) Co; (d) Pt
2.2 电化学性能表征
图4(a)为NaBH4的添加量对于DN-Pt2.8Co/C的ECSA的影响。当NaBH4的添加量控制在100 mg时, Pt2.8Co/C催化剂的ECSA达到最大值51.6 m2/gPt, 与TKK商用催化剂(28.1 m2/gPt)相比明显提高。这是因为NaBH4添加量会影响Pt晶核的生长, 从而改变催化剂的ECSA。适量的NaBH4可以有效减少由PtCl62-阴离子团还原的Pt晶核间的团聚, 阻碍其它PtCl62-在Pt晶核上的吸附, 从而限制晶核的继续生长, 避免催化剂的颗粒尺寸过大。
图4
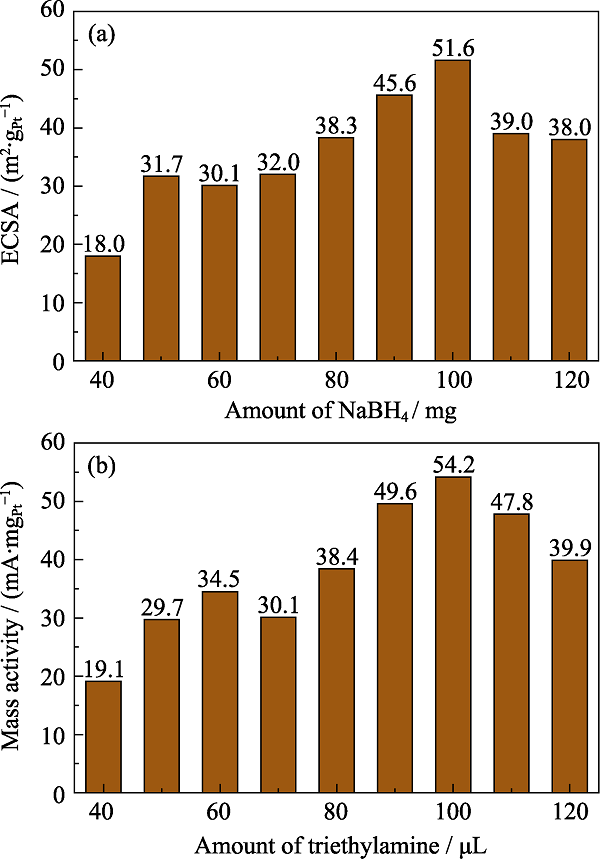
图4
不同NaBH4添加量所制备的DN-Pt2.8Co/C的ECSA直方图(a); 不同三乙胺添加量所制备DTL-Pt2.8Co/C的MA直方图(b)
Fig. 4
ECSA histogram for DN-Pt2.8Co/C with different amounts of NaBH4 (a); MA histogram for DTL-Pt2.8Co/C with different amounts of triethylamine (b)
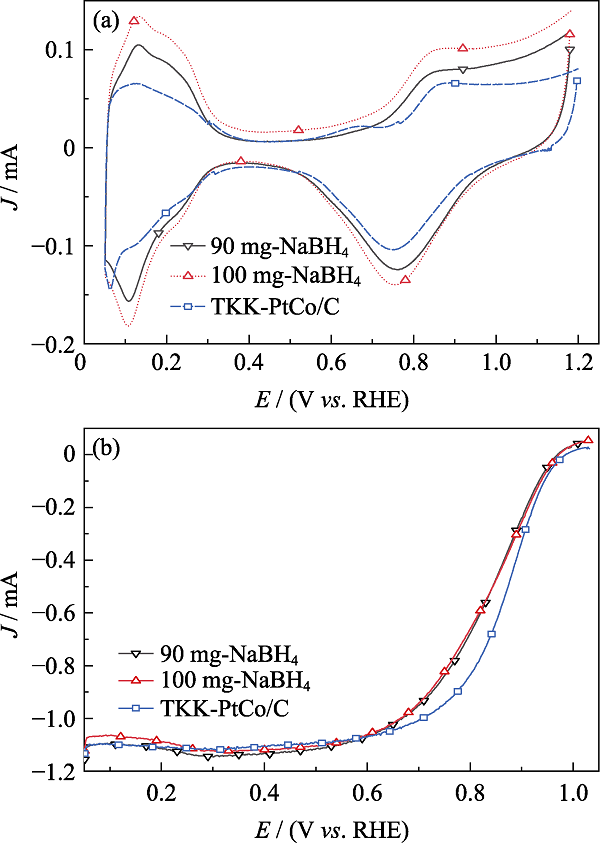
由CV曲线(图S2(a))可知, NaBH4添加量分别为90、100 mg时所制备的DN-Pt2.8Co/C氢脱附区域面积都较大, 说明其脱吸附氢原子的能力强于TKK-PtCo/C[21]。由图S2(b)中LSV曲线可知, DN-Pt2.8Co/C催化剂的半波电位均低于TKK-PtCo/C。
通过对NaBH4的调控所制备催化剂的ECSA 虽得到提升, 但合成的催化剂并未表现出较强的ORR能力。根据Yang等[22]研究, 在前驱液中添加三乙胺有助于提升ORR能力。故实验探究了三乙胺添加量对于催化剂电化学性能的影响。
图4(b)为添加不同三乙胺制备的DTL-Pt2.8Co/C催化剂的MA。当加入三乙胺量为90~110 μL时, 所制备的DTL-Pt2.8Co/C的MA有明显提升。如 图S3所示, 当三乙胺的添加量为100 μL时, DTL-Pt2.8Co/C催化剂的半波电位比TKK-PtCo/C 合金催化剂更高, 通过LSV曲线计算得到DTL-Pt2.8Co/C的MA达到54.2 mA/mgPt, 高于TKK-PtCo/C(44.3 mA/mgPt)。加入适量的三乙胺可以提升催化剂的ORR能力, 与Yang等[22]的研究结论相符合。三乙胺可与酸进行反应, 如与氯化氢(HCl)结合成三乙胺盐酸盐[23]。在前驱液中, 三乙胺与氯铂酸反应生成有机复合络合物, 能均匀地分布在碳基上, 形成浑浊悬浮的胶体溶液, 有助于金属颗粒均一地负载在碳黑上, 从而极大影响金属颗粒的生长过程, 提升金属颗粒的电催化性能[24]。
高温热处理对Pt2.8Co/C的ECSA的影响如 图S4(a)所示。高温热处理后, Pt2.8Co/C的ECSA均有所下降, 这是因为高温使催化剂的金属颗粒尺寸变大, 比表面积下降, 催化剂的活性位点变少。当温度较高时, Pt2.8Co/C催化剂会出现烧结的现象, 使催化剂金属颗粒出现较大团聚, 催化剂的电化学活性急剧下降[25]。高温热处理对Pt2.8Co/C的MA的影响如图S4(b)所示, 高温热处理后, Pt2.8Co/C-T的MA都有明显提升, 这是由于高温热处理后, 催化剂金属颗粒变成有序结构, Pt原子和Co原子的有序配位作用及有序几何作用大大提升了ORR能力[26]。同时500 ℃为最佳热处理温度, 此温度下Pt2.8Co/C-500既无大面积的金属团聚, 具有较多的电化学活性位点, 又拥有最高的ORR性能。通过CV曲线(图5(a))的对比得出Pt2.8Co/C-500的氢脱附区域面积比Pt2.8Co/C更小, 说明Pt2.8Co/C-500催化剂吸附电荷能力比Pt2.8Co/C弱。如表1所示, 虽然Pt2.8Co/C-500的ECSA比Pt2.8Co/C低, 但仍高于TKK-PtCo/C。如图5(b)所示, 通过LSV曲线对比可知, Pt2.8Co/C-500的半波电位明显高于Pt2.8Co/C和TKK-PtCo/C。如表1所示, 其MA为133 mA/mgPt, 是TKK-PtCo/C的3倍, 结合XRD图谱(图1)和TEM照片(图2), 说明更优的结晶度、更高的合金化程度可有效提升催化剂的ORR能力。
图5
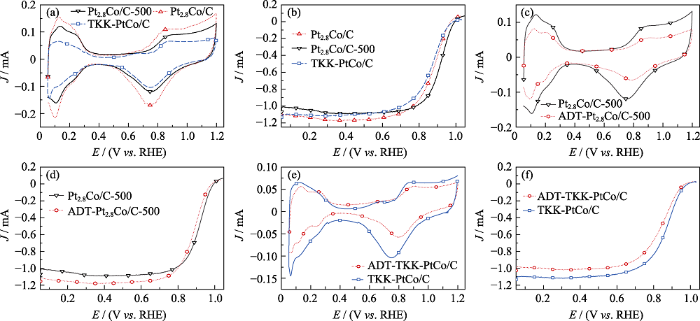
图5
Pt2.8Co/C、Pt2.8Co/C-500、TKK-PtCo/C的CV (a)和LSV曲线(b); 加速稳定性测试(ADT)前后Pt2.8Co/C-500(c, d), TKK-PtCo/C(e, f)的CV(c, e)和LSV(d, f)曲线
Fig. 5
CV(a) and LSV (b) curves for Pt2.8Co/C, Pt2.8Co/C-500 and TKK-PtCo/C; CV(c, e) and LSV(d, f) curves of Pt2.8Co/C-500 (c, d) and TKK-PtCo/C (e, f) before and after durability tests
Pt2.8Co/C-500加速稳定性测试前后的CV及LSV曲线如图5(c,d)所示。由图5(c)可得, 经过5000圈的CV循环扫描后, Pt2.8Co/C-500的氢脱附区面积减小, 这说明催化剂表面的活性位点减少, 通过CV曲线计算得出, Pt2.8Co/C-500的ECSA(表1)由43.5 m2/gPt降到24.6 m2/gPt, 降幅39.4%。由图5(d)可知, 经过5000圈CV循环扫描后, Pt2.8Co/C-500的半波电位明显减小, 负偏移34 mV, 如表1所示, MA由133 mA/mgPt降低到71 mA/mgPt, 降幅为46%。TKK-PtCo/C加速稳定性测试的CV及LSV曲线如图5(e,f)所示, 经过5000圈CV循环扫描后, TKK-PtCo/C的ECSA和MA都明显降低, 其ECSA由28.1 m2/gPt降到15.7 m2/gPt (表1), 降幅为27.5%。如图5(f)所示, 经过加速稳定性测试后, TKK-PtCo/C的半波电位负偏移23 mV, MA由44.3 mA/mgPt减小到27.2 mA/mgPt (表1), 降幅为38.6%。这说明Pt2.8Co/C-500的初始电化学性能虽然较高, 但其稳定性比TKK-PtCo/C弱。这是由于在酸性条件下, 催化剂经过加速稳定性测试后, 其金属表面的原始结构被破坏, 部分金属颗粒脱落[27,28], 导致催化剂电化学性能降低。如图S5所示, 加速稳定性测试后, Pt2.8Co/C-500电催化剂金属颗粒出现明显团聚, 其平均颗粒尺寸由3.85 nm增加到5.35 nm, 晶格间距由0.223 nm增加到0.233 nm, 这论证了其原始结构被破坏是导致电化学性能衰减的主要原因之一。此外, 碳载体结构受到电化学腐蚀的影响, 也会导致催化剂的活性位点减少、氧还原反应能力减弱[8]。
表1 PtCo合金电催化剂的ECSA和MA数据
Table 1
| Sample | ECSA/(m2∙gPt-1) | MA/(mA∙mgPt-1) |
|---|---|---|
| Pt2.8Co/C | 51.6 | 54.2 |
| Pt2.8Co/C-500 | 43.5 | 133.0 |
| TKK-PtCo/C | 28.1 | 44.3 |
| ADT-Pt2.8Co/C-500 | 24.6 | 71.0 |
| ADT-TKK-PtCo/C | 15.7 | 27.2 |
3 结论
本研究通过实验优化后确定: 添加100 mg硼氢化钠及100 μL三乙胺在500 ℃高温热处理的条件下制备的Pt2.8Co/C-500纳米合金电催化剂的电化学性能最优。与商用TKK-PtCo/C电催化剂相比, Pt2.8Co/C-500合金纳米电催化剂分散较好、合金颗粒尺寸更小且电化学活性面积更大, 催化剂活性位点更多, Pt2.8Co/C-500的ECSA达到43.5 m2/gPt, 超过商用TKK-PtCo/C电催化剂的28.1 m2/gPt; 特别在氧还原反应性能上, Pt2.8Co/C-500的质量活性达到133 mA/mgPt, 是商用TKK-PtCo/C电催化剂的3倍, 拥有更强的ORR能力。经过5000圈CV扫描后, TKK-PtCo/C的稳定性较优, Pt2.8Co/C-500的稳定性需进一步加强。本研究合成的Pt2.8Co/C-500可以提升Pt的利用率, 其更高的质量活性可减少催化剂使用量, 降低质子交换膜燃料电池的成本。
补充材料
本文相关补充材料可登陆
补充材料:
铂钴合金纳米电催化剂的制备及性能研究
朱 勇1, 顾 军1,2, 于 涛1,3, 何海佟1, 姚 睿1
(1. 南京大学 物理学院, 南京 210093; 2. 南京东焱氢能源科技有限公司, 南京 211100; 3. 南京大学 固体微结构物理国家重点实验室, 南京 210093)
图S1
图S2
图S2
不同NaBH4添加量所制备的DN-Pt2.8Co/C和TKK-PtCo/C合金催化剂的CV(a)和LSV(b)曲线
Fig. S2
CV(a) and LSV(b) curves of TKK-PtCo/C and DN-Pt2.8Co/C
图S3
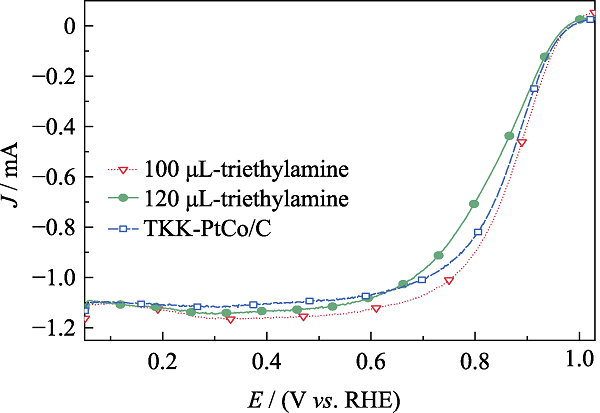
图S3
不同三乙胺添加量所制备的DTL-Pt2.8Co/C的LSV曲线
Fig. S3
LSV curves of DTL-Pt2.8Co/C with different triethylamine
图S4
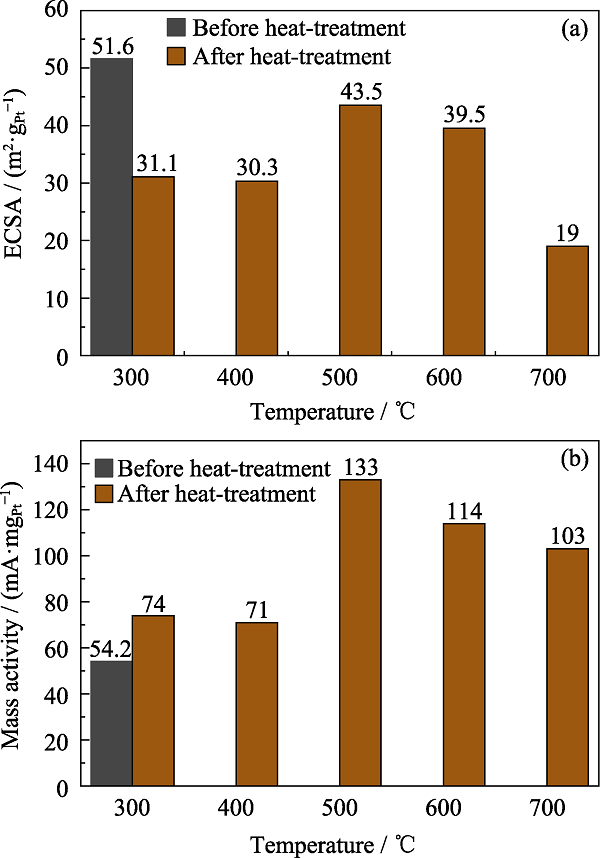
图S4
高温热处理前后的Pt2.8Co/C电化学活性面积(a)和质量活性(b)直方图
Fig. S4
ECSA (a) and MA (b) histograms of Pt2.8Co/C with different heat-treatment temperatures
图S5

图S5
ADT-Pt2.8Co/C-500的TEM照片及粒径分布图=S
Fig. S5
TEM images and particle size distribution of ADT-Pt2.8Co/C-500= S
参考文献
A comprehensive review of Pt electrocatalysts for the oxygen reduction reaction: nanostructure, activity, mechanism and carbon support in PEM fuel cells
Combined electron and structure manipulation on Fe-containing N-doped carbon nanotubes to boost bifunctional oxygen electrocatalysis
Active sites engineering of Pt/CNT oxygen reduction catalysts by atomic layer deposition
Study on durability of Pt supported on graphitized carbon under simulated start-up/shut-down conditions for polymer electrolyte membrane fuel cells
High performance Fe- and N- doped carbon catalyst with graphene structure for oxygen reduction
Research progress on advanced carbon materials as Pt support for proton exchange membrane fuel cells
Confining ultrasmall bimetal alloys in porous N-carbon as scalable and sustainable electrocatalysts for rechargeable Zn-air batteries
Alloyed Pt3 M (M=Co, Ni) nanoparticles supported on S- and N-doped carbon nanotubes for the oxygen reduction reaction
Porous cobalt oxide nanoplates enriched with oxygen vacancies for oxygen evolution reaction
N-doped defective carbon with trace Co for efficient rechargeable liquid electrolyte-/all-solid-state Zn-air batteries
The structure and activity of Pt-Co alloys as oxygen reduction electrocatalysts
Fe-PPy-TsOH/C as cathode catalyst for proton exchange membrane fuel cells
Electrocatalytic enhancement of 0D/1D/2D multidimensional PtCo alloy@cobalt benzoate/graphene composite catalyst for alcohol electro-oxidation
Platinum-cobalt bimetallic nanoparticles in hollow carbon nanospheres for hydrogenolysis of 5-hydroxymethylfurfural

The synthesis of 2,5-dimethylfuran (DMF) from 5-hydroxymethylfurfural (HMF) is a highly attractive route to a renewable fuel. However, achieving high yields in this reaction is a substantial challenge. Here it is described how PtCo bimetallic nanoparticles with diameters of 3.6 +/- 0.7 nm can solve this problem. Over PtCo catalysts the conversion of HMF was 100% within 10 min and the yield to DMF reached 98% after 2 h, which substantially exceeds the best results reported in the literature. Moreover, the synthetic method can be generalized to other bimetallic nanoparticles encapsulated in hollow carbon spheres.
Potential-cycling synthesis of single platinum atoms for efficient hydrogen evolution in neutral media
Single-atom catalysts (SACs) have exhibited high activities for the hydrogen evolution reaction (HER) electrocatalysis in acidic or alkaline media, when they are used with binders on cathodes. However, to date, no SACs have been reported for the HER electrocatalysis in neutral media. We demonstrate a potential-cycling method to synthesize a catalyst comprising single Pt atoms on CoP-based nanotube arrays supported by a Ni foam, termed PtSA-NT-NF. This binder-free catalyst is centimeter-scale and scalable. It is directly used as HER cathodes, whose performances at low and high current densities in phosphate buffer solutions (pH 7.2) are comparable to and better than, respectively, those of commercial Pt/C. The Pt mass activity of PtSA-NT-NF is 4 times of that of Pt/C, and its electrocatalytic stability is also better than that of Pt/C. This work provides a large-scale production strategy for binder-free Pt SAC electrodes for efficient HER in neutral media.
Low-temperature hydrogen production from water and methanol using Pt/alpha-MoC catalysts
Polymer electrolyte membrane fuel cells (PEMFCs) running on hydrogen are attractive alternative power supplies for a range of applications, with in situ release of the required hydrogen from a stable liquid offering one way of ensuring its safe storage and transportation before use. The use of methanol is particularly interesting in this regard, because it is inexpensive and can reform itself with water to release hydrogen with a high gravimetric density of 18.8 per cent by weight. But traditional reforming of methanol steam operates at relatively high temperatures (200-350 degrees Celsius), so the focus for vehicle and portable PEMFC applications has been on aqueous-phase reforming of methanol (APRM). This method requires less energy, and the simpler and more compact device design allows direct integration into PEMFC stacks. There remains, however, the need for an efficient APRM catalyst. Here we report that platinum (Pt) atomically dispersed on alpha-molybdenum carbide (alpha-MoC) enables low-temperature (150-190 degrees Celsius), base-free hydrogen production through APRM, with an average turnover frequency reaching 18,046 moles of hydrogen per mole of platinum per hour. We attribute this exceptional hydrogen production-which far exceeds that of previously reported low-temperature APRM catalysts-to the outstanding ability of alpha-MoC to induce water dissociation, and to the fact that platinum and alpha-MoC act in synergy to activate methanol and then to reform it.
Achieving remarkable activity and durability toward oxygen reduction reaction based on ultrathin Rh-doped Pt nanowires
The research of active and sustainable electrocatalysts toward oxygen reduction reaction (ORR) is of great importance for industrial application of fuel cells. Here, we report a remarkable ORR catalyst with both excellent mass activity and durability based on sub 2 nm thick Rh-doped Pt nanowires, which combine the merits of high utilization efficiency of Pt atoms, anisotropic one-dimensional nanostructure, and doping of Rh atoms. Compared with commercial Pt/C catalyst, the Rh-doped Pt nanowires/C catalyst shows a 7.8 and 5.4-fold enhancement in mass activity and specific activity, respectively. The combination of extended X-ray absorption fine structure analysis and density functional theory calculations reveals that the compressive strain and ligand effect in Rh-doped Pt nanowires optimize the adsorption energy of hydroxyl and in turn enhance the specific activity. Moreover, even after 10000 cycles of accelerated durability test in O2 condition, the Rh-doped Pt nanowires/C catalyst exhibits a drop of 9.2% in mass activity, against a big decrease of 72.3% for commercial Pt/C. The improved durability can be rationalized by the increased vacancy formation energy of Pt atoms for Rh-doped Pt nanowires.
Lattice-strain control of the activity in dealloyed core-shell fuel cell catalysts
Electrocatalysis will play a key role in future energy conversion and storage technologies, such as water electrolysers, fuel cells and metal-air batteries. Molecular interactions between chemical reactants and the catalytic surface control the activity and efficiency, and hence need to be optimized; however, generalized experimental strategies to do so are scarce. Here we show how lattice strain can be used experimentally to tune the catalytic activity of dealloyed bimetallic nanoparticles for the oxygen-reduction reaction, a key barrier to the application of fuel cells and metal-air batteries. We demonstrate the core-shell structure of the catalyst and clarify the mechanistic origin of its activity. The platinum-rich shell exhibits compressive strain, which results in a shift of the electronic band structure of platinum and weakening chemisorption of oxygenated species. We combine synthesis, measurements and an understanding of strain from theory to generate a reactivity-strain relationship that provides guidelines for tuning electrocatalytic activity.
Monodispersed Pt3Ni nanoparticles as a highly efficient electrocatalyst for PEMFCs
Pt/C catalysts with narrow size distribution prepared by colloidal-precipitation method for methanol electrooxidation
Structural parameters of supported fuel cell catalysts: the effect of particle size, inter-particle distance and metal loading on catalytic activity and fuel cell performance
Temperature- dependence of oxygen reduction activity on Pt/C and PtCr/C electrocatalysts synthesized from microwave-heated diethylene glycol method
Beneficial role of copper in the enhancement of durability of ordered intermetallic PtFeCu catalyst for electrocatalytic oxygen reduction
Platinum- iron-nickel trimetallic catalyst with superlattice structure for enhanced oxygen reduction activity and durability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}