
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CO在U2N3+x Oy 薄膜表面反应特性研究
[罗丽珠1  , 陆雷
, 陆雷2 , 刘柯钊1 , 赵东海1 ]
, 陆雷|
|


作者简介: 罗丽珠(1980-), 女, 硕士. E-mail:leezluo@163.com
采用磁控溅射沉积方法在Si基底表面制备U2N3+
U2N3+
铀氮化物具有良好的抗腐蚀性能, 并且与铀基底应力匹配良好, 被认为是重要的铀及合金表面改性层。改性过程中, 工艺条件的差异会导致薄膜组分和结构的差异。环境中氧、碳等可能会导致含氧或含碳氮化物的生成。 而含氧或含碳铀氮化物的表面腐蚀行为研究鲜有报道[ 1, 2]。作为铀氮化物的中间氧化产物UN xO y或U2N3O y[ 3, 4], 研究其腐蚀行为将有利于深入认识铀氮化物的腐蚀机理。文献研究表明: CO气氛在清洁金属铀表面吸附解离, 并生成铀的碳化物和少量自由碳[ 5, 6, 7]; 而对于金属铀表面氧化层而言, CO又能抑制其进一步氧化[ 8, 9], 这在固体表面反应中比较罕见。本工作通过磁控溅射沉积的方法在硅基底表面沉积U2N3+ xO y薄膜, 采用X光电子能谱(XPS)原位分析技术观测U2N3+ xO y薄膜在CO气氛环境下的表面腐蚀行为, 并对其反应机理进行探讨。
采用磁控溅射沉积法在Si基底表面沉积铀氮氧化物, 沉积室本底真空优于1×10-6Pa, 沉积过程中N2+Ar混合气体分压保持在10 Pa左右, 沉积时间30 min。XPS分析在ESCALAB 250型X光电子能谱仪上进行。实验过程中采用单色化Al Kα(1486.6 eV)作为激发光源, 分析室本底真空度优于1×10-7Pa, 用Ag3d5/2峰(368.26 eV)标定能谱仪, 系统分辨率为0.6 eV; 利用1 kV、1 μA氩离子枪进行薄膜深度剖析, 实验采用污染碳C1s峰结合能为285.0 eV校准图谱。在XPS能谱仪的分析室中进行薄膜的原位反应实验, 氧化过程中微漏阀控制反应气体CO(纯度优于99.999%)分压至~5.5×10-6Pa, 并通过反应时间控制气氛反应量。
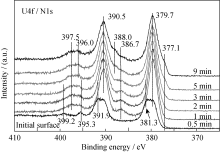
沉积的薄膜在大气环境中放置约一周, 然后对样品表面状态进行分析, 表面有一定程度氧化。图1和图2是薄膜表面U4f、N1s和O1s谱随溅射时间的变化情况。经过溅射, U4f主峰峰位和强度没有明显变化, 但卫星峰的变化相对复杂。溅射0.5 min后, U4f7/2和U4f5/2特征峰对应的结合能位置分别为379.7 eV和390.5 eV; 但分别在386.7和397.5 eV处出现两个卫星峰。该峰结合能位置高于U2N3(378.4 eV)[ 10], 且具有良好的对称性, 说明该物质比U2N3具有更高的化学态。文献[2, 11]未观察到类似卫星峰的存在, 而Allen等[ 12]在实验结果中观察到类似的现象, Black充分证明该物质应为U2N3+ x[ 10]。由此认为386.7和397.5 eV处两卫星峰对应的物质可能是U2N3+ x或含有少量间隙氧的U2N3+ x1O y1(当 y=0, 即为U2N3+ x,)。为便于与薄膜主体成分的对比区分, 下文用“富氮层”来描述U2N3+ x或含有少量间隙氧的U2N3+ x1O y1。
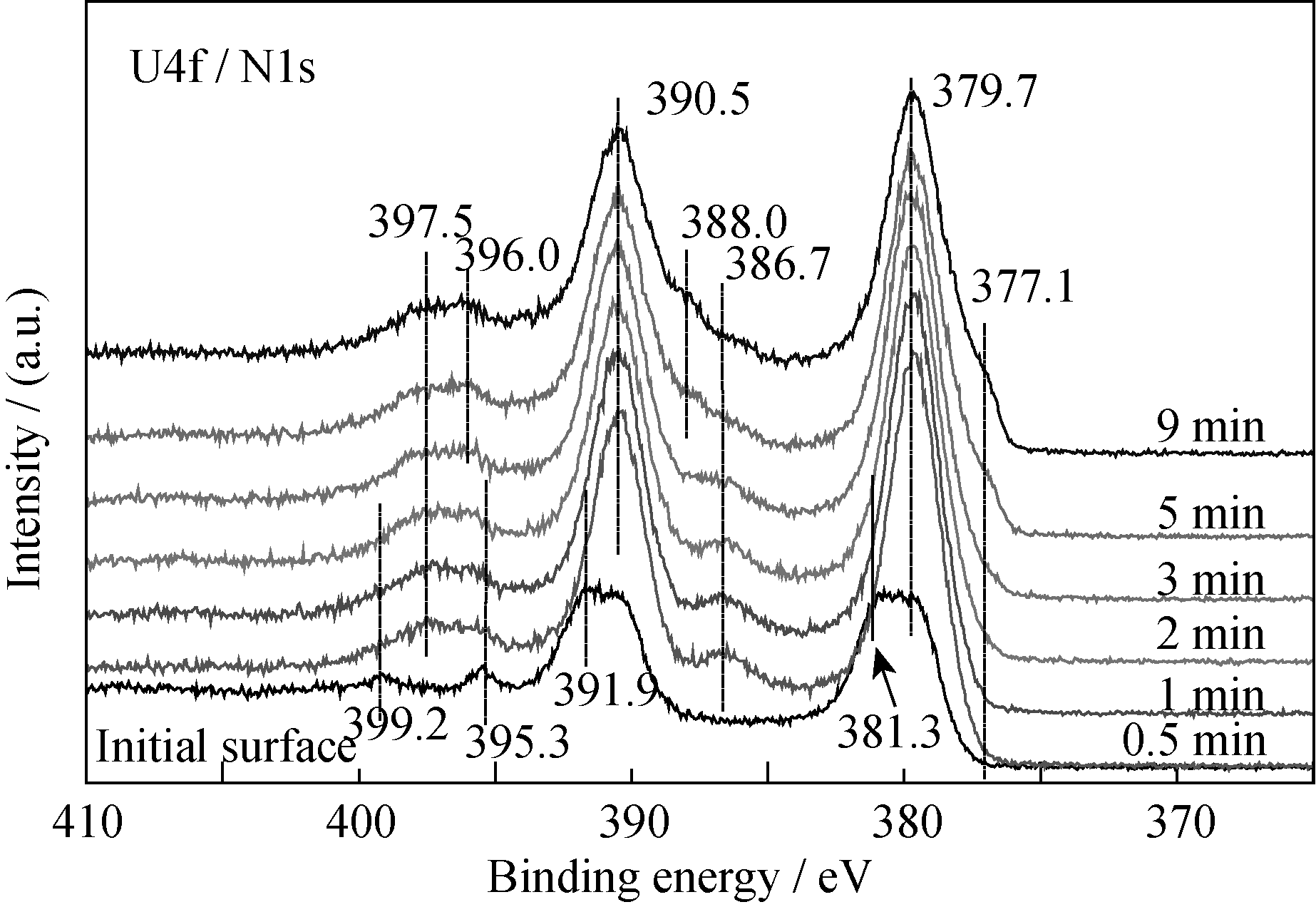 | 图1 U4f和N1s谱随溅射时间的变化Fig. 1 Spectra of U4f and N1s as function of sputtering time |
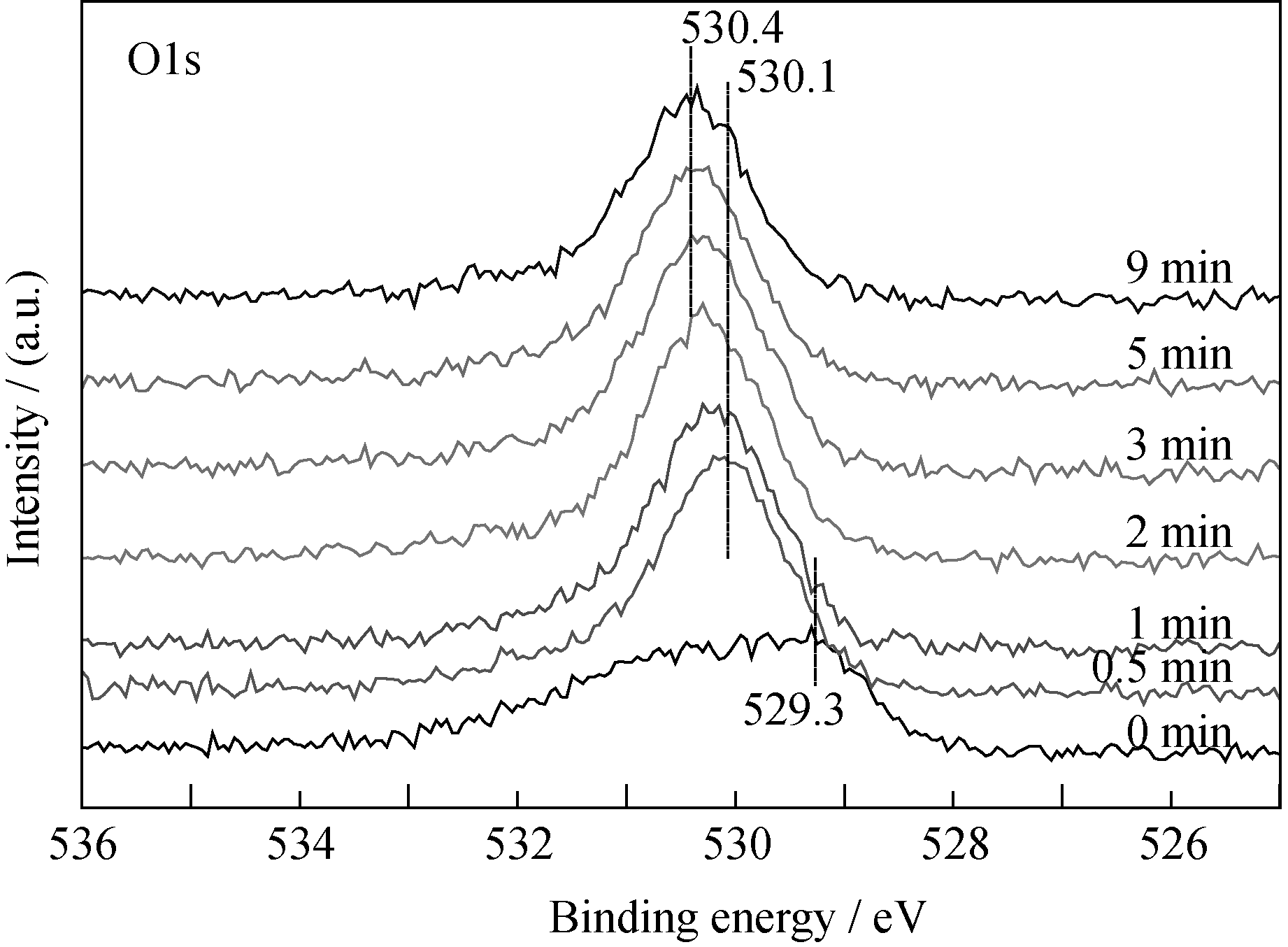 | 图2 O1s谱随溅射时间的变化Fig. 2 Spectra of O1s as function of sputtering time |
随着溅射时间的延长, 两个卫星峰强度先增强, 后逐渐减弱, 主峰强度也发生相应的变化, 即: 在溅射2 min时, 薄膜表面主峰和卫星峰强度都达到最大值, 随后逐渐减弱。由此说明富氮层组分的主峰位置与薄膜主体组分的U4f结合能位置相近, U4f7/2和U4f5/2峰位分别为~379.7 eV和390.5 eV, 且富氮层沿薄膜深度分布逐渐减少。溅射5 min后, 分别在377.1和388.0 eV处出现凸肩, 主要来源于UN[ 12], 可能是由于Ar离子对U2N3+ xO y薄膜中非金属元素的择优溅射引起的。
结合O1s谱的变化情况可知, 在工艺参数恒定的条件下沉积所得的薄膜是均一的铀氮氧化物, XRD分析结果(图略)也证明了这一点。但是文献[1-2]并未观察到类似的卫星峰结构, UN xO y是UO和UN的固溶体, 其中第二相为满足晶格匹配而在主相化合物中合理分布。根据XPS半定量分析认为, 沉积所得的薄膜主要组分为U2N3+ xO y, 其中 x< x1, y> y1; 其结构可能是氧以间隙离子形式存在于U2N3+ x晶格中, 也可能是UO与U2N3+ x的固溶体。
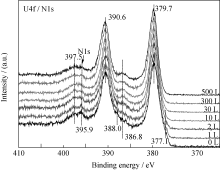
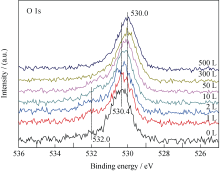
U2N3+ xO y薄膜与CO反应过程中, U4f谱卫星峰(386.7、397.5 eV)强度变化(图3)与深度剖析过程(图1)相似, U4f主峰的半高宽(FWHM)先减小后增大。 当CO暴露量为10 L时, 样品表面U4f峰及卫星峰强度最大, 此后, U4f谱峰逐渐减弱; 而O1s谱则随CO暴露量逐渐向低能端偏移且强度增大, 如图4所示。 当CO暴露量小于2 L时, O1s谱在~532 eV附近有一个肩峰, 对应的薄膜表面吸附的CO。O1s谱的变化说明CO反应过程中, 样品表面O/U比逐渐增大, 即生成了更高价的铀氮氧化物或/和铀氧化物。
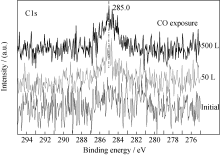
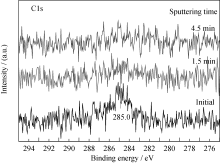
反应过程中, 样品表面探测到一定量碳的信号(图5), 其对应的结合能位置为285.0 eV, 为无定形碳, 它可能来源于CO分子在薄膜表面的吸附解离; 而CO暴露反应后的深度剖析过程中并未观测到C1s信号(图6), 说明反应过程中无定形碳主要聚集在薄膜表面, 而未观察到碳向薄膜内层的扩散, 该现象不同于金属铀与CO的反应[ 5, 6, 7]。受反应过程中CO暴露量的限制, N1s峰(395.9 eV)并未发生明显的化学位移, 但强度一定程度减弱。
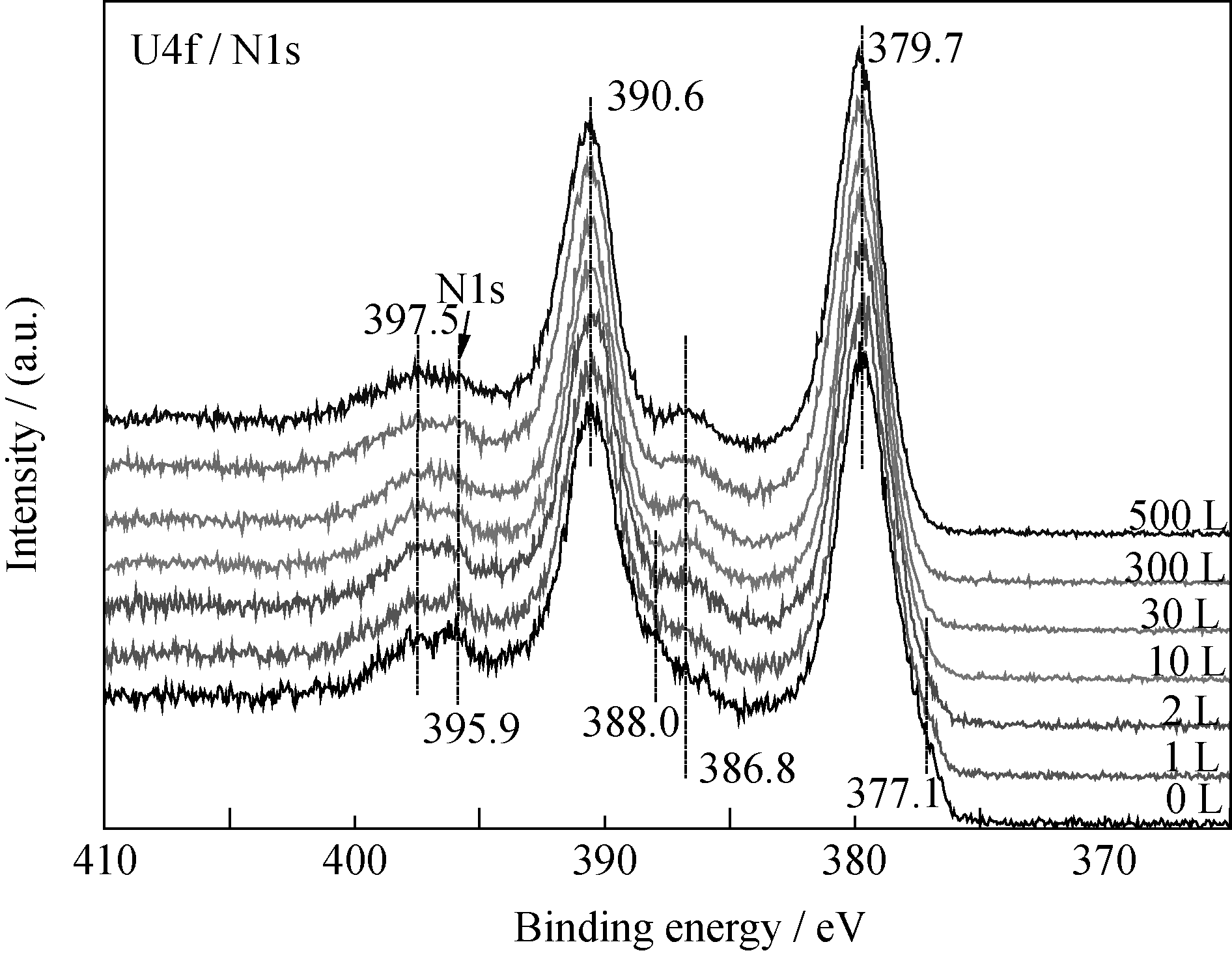 | 图3 U4f、N1s谱随CO暴露量的变化情况Fig. 3 Spectra of U4f and N1s as function of CO exposure |
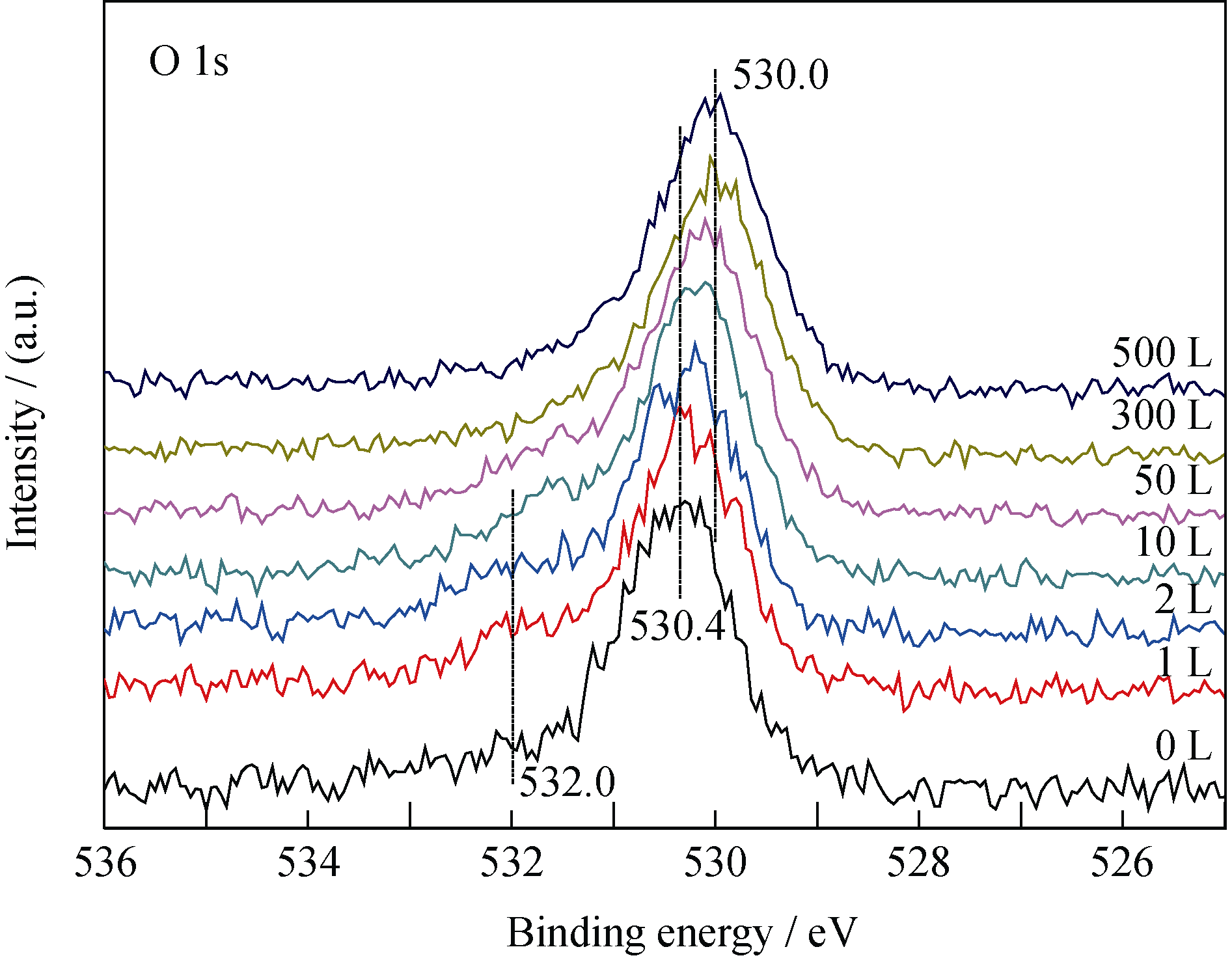 | 图4 O1s谱随CO暴露量的变化情况Fig. 4 Spectra of O1s as function of CO exposure |
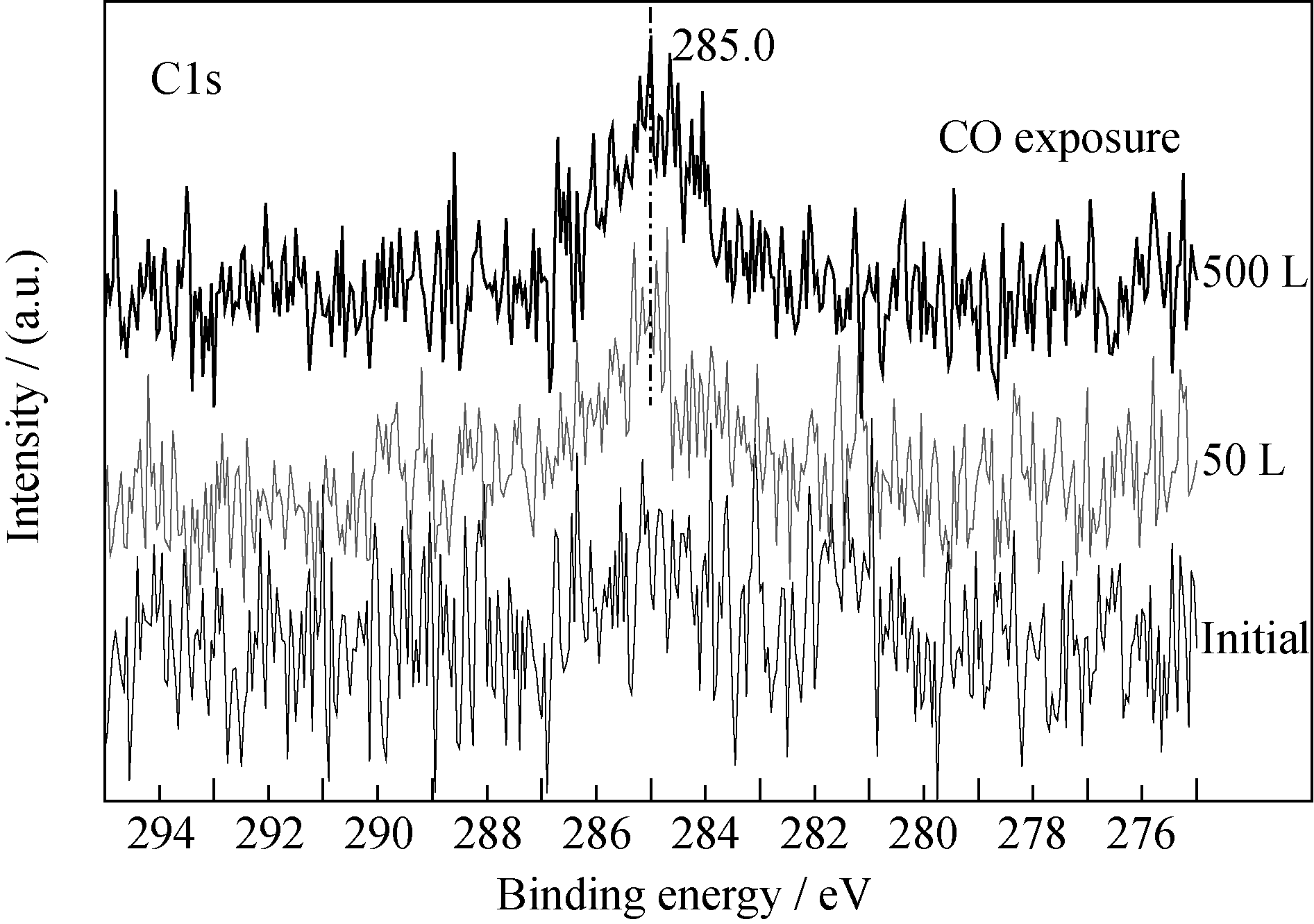 | 图5 C1s谱随CO暴露量的变化情况Fig. 5 Spectra of C1s as function of CO exposure |
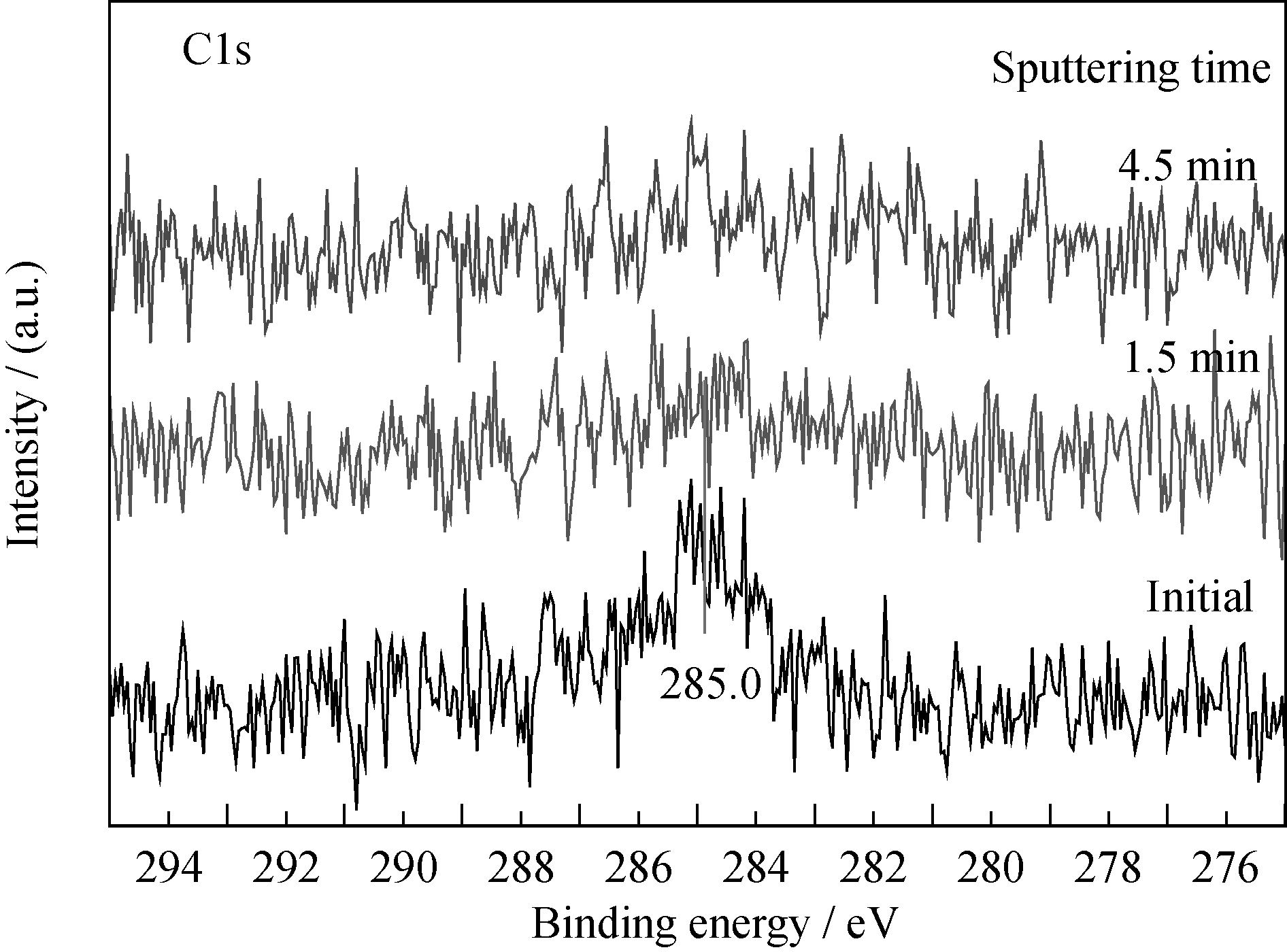 | 图6 CO暴露反应后C1s谱随溅射时间的变化情况Fig. 6 Spectra of C1s as function of sputtering time after reaction with CO |
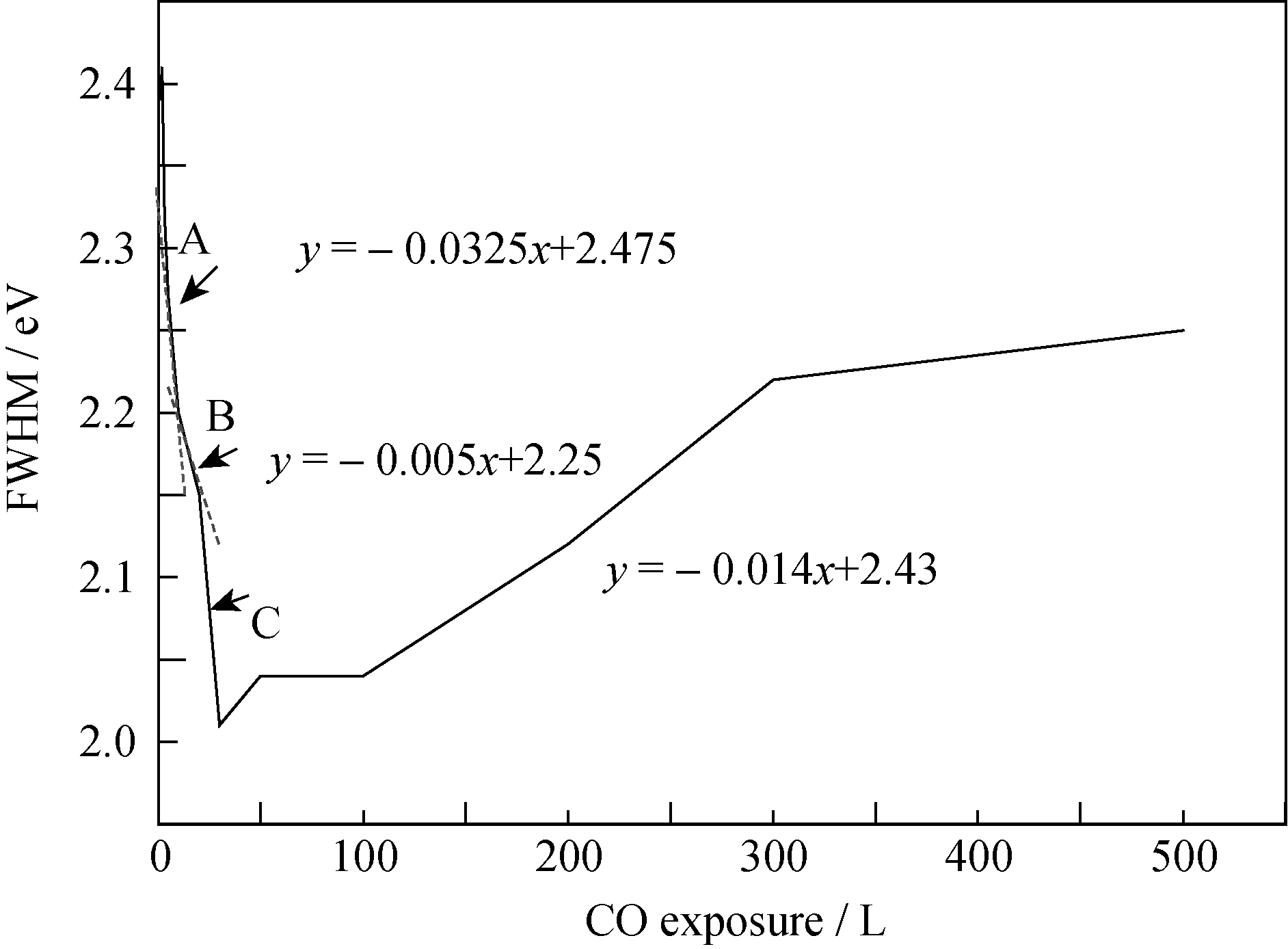 | 图7 U4f7/2峰半高宽随CO暴露量的变化情况Fig. 7 Variation of FWHM of U4f7/2 peaks as function of CO exposure |
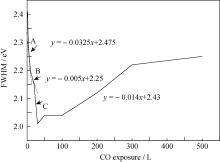
为了进一步认识CO在UNO薄膜表面的腐蚀行为, 实验对U4f谱峰半高宽变化进行了系统分析。 当CO暴露量为0~30 L时, U4f7/2峰半高宽变化非单调减小, 分别用三个函数对三个不同阶段(A、B、C)进行描述, 如图7所示。当CO暴露量为0~10 L时(阶段A), U4f7/2峰半高宽迅速减小; 当CO暴露量为10~20 L(阶段B)时, 半高宽变化逐渐减缓; 而当CO暴露量为20~30 L时(阶段C), 半高宽变化介于阶段A和B之间。
反应初期, O1s谱出现~532.0 eV处的肩峰, 说明CO在样品表面吸附解离; 随后, U4f谱中377.1和388.0 eV处凸肩消失、主峰强度增大, 说明样品表面的UN被氧化生成高价的UN xO y, 所得U4f峰较UN向高能端偏移, 并与U2N3+ xO y发生重叠; 而卫星峰强度递增意味着表面探测到的富氮层组分U2N3+ x1O y1含量增多, 并在CO暴露量为10 L时达到最大值。二者是导致阶段A谱峰半高宽减小的主要原因, 其可能的化学反应为:
COads.→Cads.+Oads. (1)
UN+Oads.→UN xO y(inst.) (2)
U2N3+ xO y+Oads.→U2N3+ x2O y2+N ( x2< x, y2> y) (3)
U2N3+ xO y+N→U2N3+ x1 O y1+O( x1> x, y1< y) (4)
随着CO暴露量的增加(阶段B), 样品表面的U2N3+ xO y及富氮层的U2N3+ x1O y1被继续氧化(反应3和反应5), 导致卫星峰强度的减弱。
U2N3+ x1O y1+Oads.→U2N3+ x1O y +N (5)
在CO环境下, 薄膜表面反应(3)将持续进行, 从而推进反应(4)和(5)的不断发生。阶段C卫星峰强度的减弱及U4f7/2峰半高宽的变窄说明表面组分逐渐单一化, 也就是说样品表面探测到的富氮层U2N3+ x1O y1减少, 表面组分主要为U2N3+ xO y。
当薄膜表面进一步氧化, 将生成高价的铀氧化物或铀氮氧化物, 从而导致了U4f峰向高能端展宽:
U2N3+ xO y+Oads.→UO2+ x+N (6)
随着样品表面高价氧化物或铀氮氧化物的生成, 样品表面解离的O在薄膜的扩散受到抑制, 使得薄膜表面氧化速率降低, 使得U4f谱的展宽变缓。
UN表面氧化过程中出现包含U2N3的三明治结构UO2/U2N3/UN[ 1, 13, 14, 15], 而本研究也观测到类似的结构: U2N3+ xO y/U2N3+ x1O y1 /U2N3+ xO y+UN。根据U4f谱变化可知: U2N3+ xO y与CO反应过程中也会生成中间产物, 形成富氮层(U2N3+ x1O y1)。结合XPS分析结果可以认为: CO反应及反应后深度剖析过程中相似的卫星峰变化, 主要来源于富氮层中间产物的位置变化, 即氧化过程中富氮层中间产物逐渐向薄膜内部迁移, 逐步远离XPS的分析深度。
根据实验结果可得: CO在U2N3+ xO y薄膜表面表现为氧化特性; 外表面氧化反应生成的N向内部扩散, 并在次表面生成富氮的中间产物U2N3+ x1O y1; 且随CO暴露量的逐渐增多, 氧扩散为反应的主导机制, 富氮层逐渐向薄膜内层迁移。
超高真空环境下, CO在U2N3+ xO y薄膜表面表现为明显的氧化特性, 其主要反应过程为: CO在U2N3+ xO y薄膜表面的吸附解离生成的碳以无定形碳形式聚集在薄膜表面; 而解离的氧与薄膜发生氧化反应生成更高价的铀氮氧化物和/或铀氧化物; 释放的氮向薄膜内层扩散, 并在界面处生成富氮的中间产物U2N3+ x1O y1。随着反应进程推进, 富氮层逐渐向远离薄膜外表面方向迁移, 从而导致U4f卫星峰强度发生变化。当薄膜表面生成稳定的高价铀氧化物或铀氮氧化物后, 氧扩散被抑制, 氧化反应进程变缓。当然, 有限的研究还不能完全获得U2N3+ xO y的氧化机理, 还需要进一步设计实验进行深入研究。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|