{kind=link}
{kind=link}
{kind=link}
Pt和Au修饰锐钛矿型TiO2(101)面的第一性原理研究
[马新国1, 2  , 严杰
, 严杰1 , 陈紫梦1 , 祝林1 , 徐国旺1, 2 , 黄楚云1, 2 , 吕辉1, 2 ]
, 严杰, 吕辉|
|


作者简介: 马新国(1978-), 男, 博士, 副教授. E-mail: maxg2013@sohu.com
采用平面波超软赝势方法研究了Pt和Au修饰锐钛矿型TiO2(101)面的结构稳定性及电子结构。结果显示贵金属原子在TiO2(101)符合化学计量比的条件下, 在其表面的吸附作用不强, 对电子结构的影响也较小。但是发现在富O条件下, Pt和Au原子容易吸附在表面Ti空位的位置, 与Au原子不同, Pt原子有从TiO2表面扩散进入体相晶格中的趋势。而在富Ti条件下, Pt和Au原子容易吸附在O1空位的位置。对可能存在的几种空位缺陷吸附模型进行了电子结构的计算。结果表明: 空位缺陷的产生不仅有利于Pt和Au原子“湿化”TiO2(101)表面, 也有利于带隙中产生贵金属原子的5d杂质能级。
Structure stability and electronic structures of anatase TiO2(101) surface modified by noble metal Pt and Au have been investigated using plane-wave ultrasoft pseudopotentials. The results show that the adsorption interaction is weak between noble atoms and anatase TiO2(101) surface, resulting in a slight effect on its electronic structure. However, under O-rich condition, it is found that Pt and Au atoms are favor of the Ti vacancies. Contrary to Au atom, Pt atom tends to diffuse from surface into bulk. Under Ti-rich condition, Pt and Au atoms are favor of the O vacancies. The calculated electronic structures of possible vacancy defects indicate that the appearance of surface vacancies not only avails to wet anatase TiO2(101) surfaces, but also enables their atoms to appear 5d impurity energy levels in band gap.
半导体TiO2由于结构稳定、价格低廉和无毒等特点, 成为目前值得期待的光催化材料[1, 2]。但是过宽的带隙以及量子产率不高等不易克服的缺点限制了其广泛应用。响应光子的激发而形成的电子空穴对向晶体表面扩散并与表面吸附的O2、H2O反应形成强氧化性自由基的过程中, 与之相竞争的是电子-空穴对的复合, 抑制电子-空穴对复合的关键是要控制电子和空穴接触几率, 提高载流子的迁移寿命[3]。研究表明锐钛矿型TiO2纳米晶表面氧空位的存在, 一定程度上可以抑制电子和空穴的复合, 但这种抑制效率并不高。采用贵金属修饰TiO2表面成为调制表面能带结构和载流子迁移寿命的一种有效方法[4, 5, 6, 7, 8, 9]。金属的Fermi能级低于TiO2, 被光照激发后, 电子从TiO2向金属上扩散(转移), 直到两者Fermi能级相同。载流子的重新分布使电子富积在金属上, 金属和半导体界面上形成Schottky势垒, 作为电子捕获陷阱抑制电子和空穴的复合, 从而提高催化剂的活性。Coleman等[5]较早采用了Pt和Ag负载TiO2光催化材料降解水中的激素类有机物。随后, Kozlova等[6]和Li等[7]分别研究了Pt和Ru等贵金属负载TiO2的制备工艺, 但是其微结构及贵金属对表面性质影响并不清楚。
Mete等[8, 9]采用离散傅里叶变换(DFT)计算法研究了Pt和Au修饰TiO2(001)表面的电子结构和稳定性, 认为Pt和Au修饰使TiO2(001)表面金属化。锐钛矿型TiO2暴露在外的主要表面是(101)面(占94%以上), 具有表面能低、结构稳定等特点, 在可能发生的化学及光催化过程中起着至关重要的作用[10, 11]。为此, 大家把研究目标转向金属修饰TiO2(101)表面的几何结构和性质研究。Má rquez等[10]研究了W掺杂TiO2(101)表面的几何结构和电子结构, 认为W6+更可能吸附在表面Ti空位的位置, 但是对能带的调制作用十分有限, 仅减小带隙0.1~0.2 eV。Perkas等[12]也发现Pt进入TiO2晶格十分困难, 而Au要容易的多。然而Zhang等[11]的研究却发现在氧化性气氛下中性Pt原子能较容易地扩散进入TiO2晶格, 当这些Pt原子形成Pt2+时, 能替代Ti4+原子。由于获得符合实验要求的贵金属修饰的特定锐钛矿型TiO2表面微结构比较困难, 以及实验技术本身的限制, 导致上述研究均未能揭示Pt和Au修饰锐钛矿型TiO2(101)表面的点缺陷, 其对TiO2(101)表面结构和性质的影响至今不明。
之前, 我们将第一性原理平面波超软赝势方法用于研究材料表面的原子结构及电子结构的计算, 获得了较好的结果[13, 14]。本研究将采用同样的计算方法研究Pt和Au贵金属修饰锐钛矿型TiO2(101)面结构的稳定性和电子结构, 以期获得实验技术所不能提供的数据, 为进一步探讨贵金属修饰TiO2表面的光催化活性机理打下基础。
采用了基于密度泛函理论的平面波超软赝势方法[15]。在之前的锐钛矿型TiO2的几何结构研究中[16], 发现交换关联能采用广义梯度近似(GGA)中PW-91方案计算出的结果与实验吻合较好[14]。在描述离子实与价电子之间的相互作用时, 选取的价电子组态分别为O: 2s22p4, Ti: 3s23p63d24s2, Pt: 5d96s1, Au: 5d106s1, 其他轨道电子视为芯电子进行计算。为了提高计算精度, 平面波截断能Ecut取为380 eV。自洽场运算中, 应用了Pulay密度混合法, 自洽精度设为每个原子能量收敛至2.0× 10-6 eV, 第一布里渊区按5× 5× 1进行分格, 能达到收敛性要求。对模型的结构优化中, 采用了BFGS算法, 每个原子能量收敛至2.0× 10-5 eV以内, 每个原子受力不超过0.5 eV/nm, 原子的最大位移不超过0.02 nm。为了节约计算时间, 对表面第6层下的原子进行了固定, 仅仅让表面的部分原子弛豫。后面的所有计算均采用相同的设置, 通过CASTEP软件计算完成[17]。
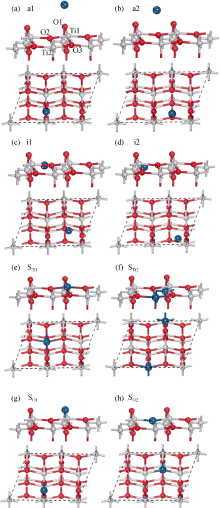
根据文献[14]中锐钛矿型TiO2(101)表面模型, 即选取真空厚度为0.8 nm, 以保证上下两层表面之间的作用可以忽略, 表面厚度选取18层原子, 其清洁表面的表面能收敛度小于0.02 J/m2。分别建立了Pt和Au吸附在表面上方、表面间隙位置以及表面原子空位位置三类结构共8种模型。表面上方的吸附模型有, a1: 金属原子吸附在O1原子上方, a2: O1与O2之间上方; 表面间隙模型有, i1: 在O1和O2之间, i2: 在O1和O3之间; 吸附在表面空位模型(Pt和Au原子取代表面Ti或者O原子)有STi1: 占据Ti1空位, STi2: 占据Ti2空位, SO1: 占据O1空位, SO2: 占据O2空位。这里分别建立了1× 1和1× 2表面模型, 其中1× 2表面吸附模型如图1所示。
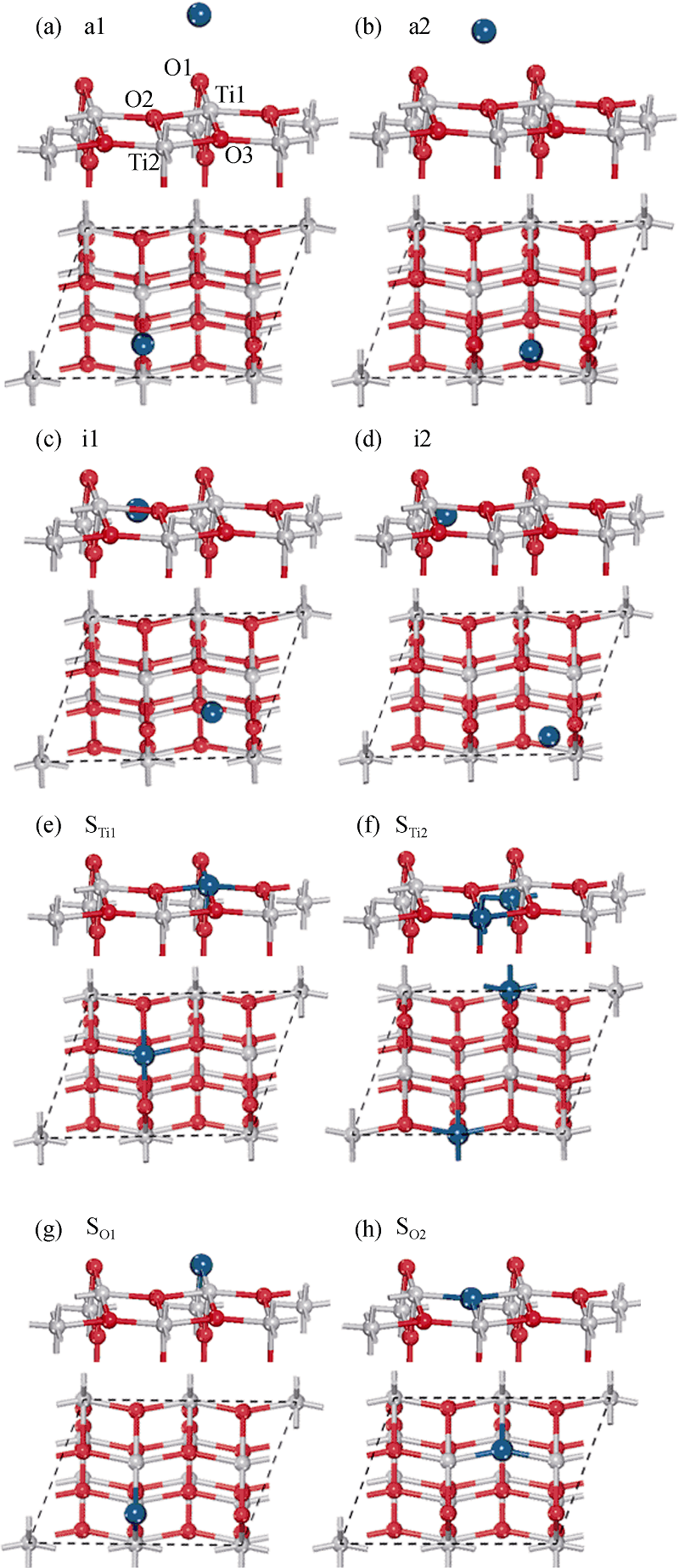 | 图1 贵金属原子吸附于锐钛矿型TiO2(101)面的侧视(上)和俯视(下)结构图。((a)和(b)是表面上方的吸附, (c)和(d)是表面间隙位置的吸附, (e)和(f)是吸附于表面Ti空位, (g)和(h)吸附于表面O空位。其中小红球和小灰球分别表示O和Ti原子, 大蓝球表示吸附的贵金属原子)Fig. 1 The structure model of anatase TiO2(101) surface modified noble metal from side and top view((a) and (b) indicate the adsorption above surface; (c) and (d) indicate the adsorption between surface atoms; (e) and (f) indicate the adsorption at Ti vacancy; (g) and (h) indicate the adsorption at O vacancy. Small black and gray spheres represent the O and Ti atoms, respectively. Big blue spheres represent the noble metal atoms) |
为了分析贵金属修饰TiO2表面几何结构的稳定性, 计算了Pt和Au吸附于表面的吸附能以及吸附于表面空位的杂质吸附能。以锐钛矿型TiO2(101)的1× 1表面面积A为单位面积, 可以定义单位面积A上表面吸附能和吸附于表面空位的吸附能Ef分别表示为[4, 7, 18, 19]
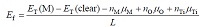
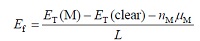
其中ET(M)为贵金属原子吸附于化学计量比表面或者空位位置时体系的总能量, ET(clear)为清洁表面结构的总能量, nM为吸附的原子数, μ M为吸附原子的绝对化学势, L为面积A的倍数。
首先采用(1)式计算了Pt和Au吸附于表面上方或者间隙位置时的吸附能。表1显示了1× 2结构中的吸附能均要低于1× 1结构, 即低浓度下的吸附能要比高浓度下的吸附能要低。可以判断在表面上方或者间隙位置的贵金属吸附浓度并不会太高。尽管在1× 1和1× 2结构中Au的吸附能均要低于Pt的吸附能, 但是这些吸附能均为正, 且都高于清洁表面的形成能, 表明贵金属在表面上方或者间隙位置的吸附作用较弱。该结论与Mete等[8, 9]研究TiO2(001)面的结果相似。文献[8]中指出在富Ti条件下, TiO2(001)面低浓度的Pt吸附最为稳定, 而且出现半导化, 其带隙窄化为1.03 eV。文献[9]中Au在该表面的吸附也表现出类似的情况。
利用(2)式计算单位面积A上Pt和Au原子分别吸附于表面Ti和O空位时的吸附能(表2), 发现当Pt吸附在表面Ti空位的位置时, 在富O条件下的吸附能比富Ti条件下的吸附能要低, 且均为负, 说明了富O条件下Pt原子优先占据Ti空位的位置。在同样的富O条件下, 高吸附浓度1× 1模型的吸附能总体要小于低吸附浓度1× 2的模型, 说明了富O条件更有利于实现Pt原子在Ti空位位置的较高浓度吸附。此外, 在1× 1和1× 2结构中均出现Pt原子吸附在Ti2空位的吸附能比Ti1空位的吸附能要低0.2~0.4 eV, 表明了Pt原子有从TiO2表面扩散进入晶格中的趋势, 这个结果与早期Perkas等[12]获得的结果有所不同, 这可能与他们采用的制备气氛等实验条件有关。当Pt原子吸附在表面O空位的位置时, 在富Ti条件下吸附能比富O条件下吸附能均要低, 而且1× 2模型的吸附能总体要小于低吸附浓度的1× 1模型。这说明在富Ti条件下Pt原子可以吸附于O空位的位置, 而且低浓度吸附情形更容易出现, 相似的结果在文献[20]也得到证实。Pt原子吸附于O1空位的吸附能比O2空位的吸附能要低, 表明在富Ti条件下Pt原子更倾向于吸附于表面O1空位的位置。
| 表1 贵金属吸附在表面上方和间隙位置的吸附能(eV) Table 1 Adsorption energies of noble metal adsorbing above surface or adsorbing between surface atoms (eV) |
| 表2 贵金属吸附在表面Ti或者O空位的吸附能(eV) Table 2 The adsorption energies of noble metal adsorbing at Ti or O vacancies in TiO2(101) surface (eV) |
当Au吸附于表面Ti空位的位置, 在富O条件下其吸附能比在富Ti条件下均要低, 说明了富O条件有利于Au吸附在表面Ti空位, 这与Pt吸附于表面Ti空位的情形类似。但不同的是Au吸附在Ti2空位的吸附能均比Ti1空位的吸附能要高, 说明了Au原子扩散进入体相晶格比较困难。在同样的富O条件下, 低吸附浓度1× 2模型的吸附能总体要小于高吸附浓度的1× 1模型, 说明了富O条件不利于实现Au原子在Ti空位的较高浓度吸附。当Au原子吸附在表面O空位的位置时, 在富Ti条件下其吸附能比在富O条件下均要低, 且1× 2模型的吸附能总体要小于高浓度吸附的1× 1模型。表明了在富Ti条件下有利于Au原子吸附在O空位的位置, 而且高浓度吸附的情形较困难。Au原子吸附于O1空位的吸附能比O2空位的吸附能要低, 表明在富Ti条件下Au原子倾向于吸附于表面O1空位的位置, 这与Pt吸附在表面O空位时的情形类似, 这种还原条件下贵金属吸附于TiO2(110)面O空位的情况, 已经较早就得到理论证实[21]。
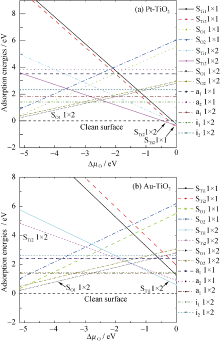
图2为吸附能与O化学势之间的关系。可以看出, 当∆ μ O=0, Pt吸附于Ti2空位的1× 1结构能量最低, 其结构最为稳定。随着∆ μ O的逐渐降低, 其吸附能逐渐增加; 当∆ μ O低于-0.135 eV, Pt吸附于Ti2空位的1× 2结构能量最低, 其结构最为稳定, 说明了随着O化学势的降低, Pt替代Ti2的浓度减少, 富O条件有利于Pt在Ti2空位的吸附。当∆ μ O低于-2.47 eV, Pt原子趋向于吸附于表面O1空位的位置。尤其可以看到, 当∆ μ O低于-0.445 eV, 没有Pt修饰的清洁表面结构最稳定。从图2(b)可以看出, 当∆ μ O=0, Au取代Ti1的1× 2结构最稳定; 随着∆ μ O的减少, 吸附能增加。当∆ μ O低于-0.88 eV, Au吸附在a1情形的1× 2结构最稳定; 当∆ μ O低于-2.70 eV, Au原子趋向吸附于表面O1空位的位置。
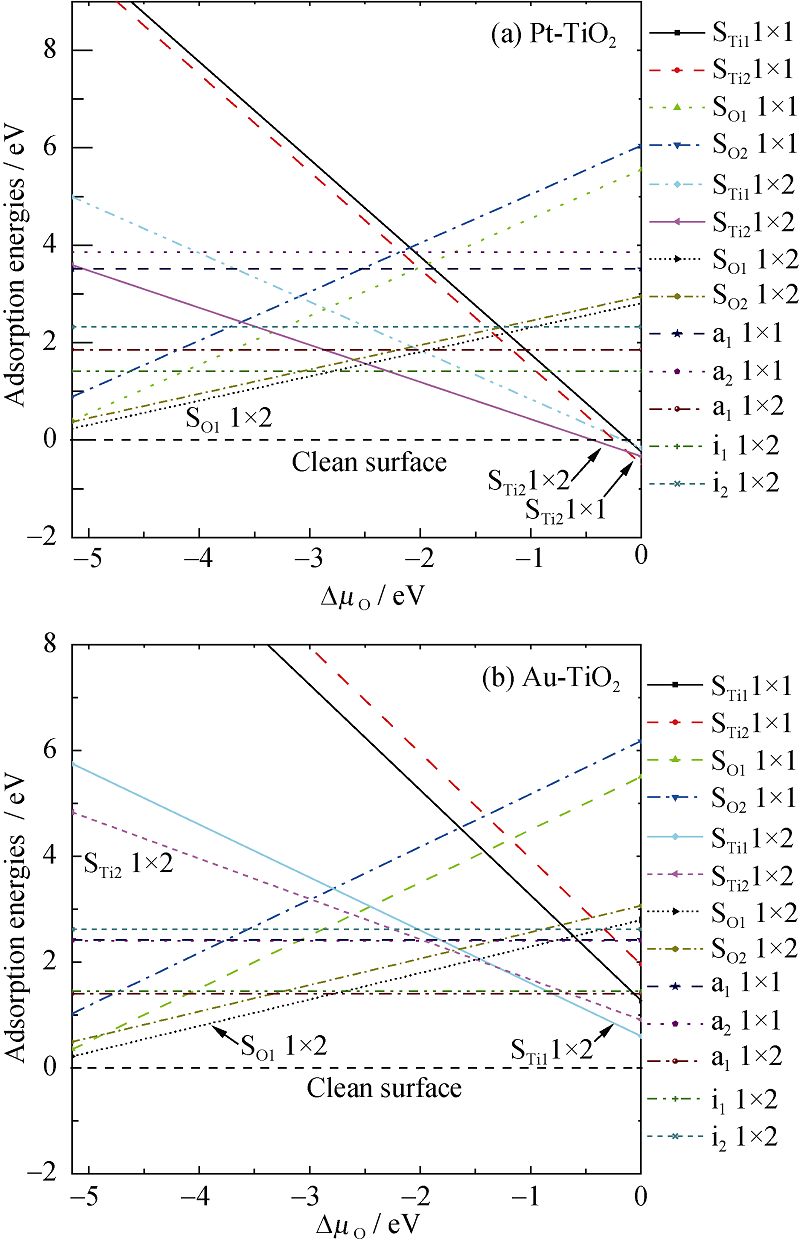 | 图2 贵金属修饰的TiO2表面结构的吸附能与O化学势之间的关系Fig. 2 The relation between the adsorption energies of TiO2(101) surface modified noble metal and the relative chemical potential ∆ μ O |
综上分析, 在O不足(富Ti)条件下将有少量的Pt优先占据O1空位的位置。随着O化学势的增强, 其吸附能也增加, Pt吸附在表面O空位的缺陷浓度越来越低, 无缺陷的清洁表面将出现。此时, 沉积在TiO2表面上的Pt原子并不会“ 湿化” 表面。之后, 随着O化学势的增强, 少量的Pt原子优先占据表面Ti2空位的位置, 或者说Pt原子优先替代Ti2原子。随着O化学势的增强, Pt占据Ti2位置的浓度逐渐增加。Au修饰TiO2表面的制备生长条件与Pt类似。不同的是在富O条件下Au原子优先占据表面的Ti1空位的位置。
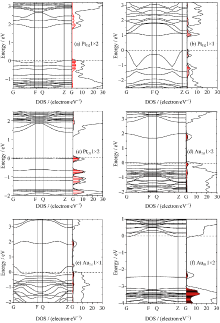
对于Pt和Au修饰表面的理论和实验研究较多, 但是贵金属的修饰对能带结构的影响仍然很少见报道, 而且对其电子结构信息的深入研究十分缺乏。为此, 针对部分可能出现的物理模型进行了能带结构和电子结构的计算。由于表面上方吸附和间隙吸附模型结构的吸附能均较高, 贵金属对TiO2表面的电子结构影响非常小[22], 为此这类模型的电子结构将在本研究中不予考虑, 这里仅计算和分析贵金属吸附在表面Ti或者O原子空位的情形。从图3可知, 贵金属修饰TiO2表面的电子结构与清洁表面有很大不同。总体上半导体表面能带的带隙中出现了许多新的能级, 它们对TiO2表面的电学性质影响很大。
图3(a)显示出Pt替代Ti2使导带底下方和价带顶上方附近分布着几条新能级, 他们在kx和ky方向上十分对称。由于Ti2原子的配位与体相结构中的情况类似, 因而在Pt替代Ti2后的电子结构与体相结构中的替代情况相似。从态密度上来看, Pt与周围O的杂化作用很强, 使部分新的缺陷能级也存在部分O2p和Ti3d的贡献。这是由于Pt的引入改变了局部的电子分布, 表面键发生重组而使能带结构中存在若干表面能带。同时在该表面上还存在O和Ti的悬挂键产生的表面能带。其中在导带底下方的能级主要是表面的空带, 这是表面不配对的悬挂键产生的, 而价带顶上方的能级是表面满带, 是表面配对的悬挂键产生的。另外, 费米能级刚好填充满价带顶上方最上的一条表面能级, 该缺陷能级到导带底下方最低缺陷能级的距离为1.81 eV, 比清洁表面的带隙约小0.8 eV[23]。
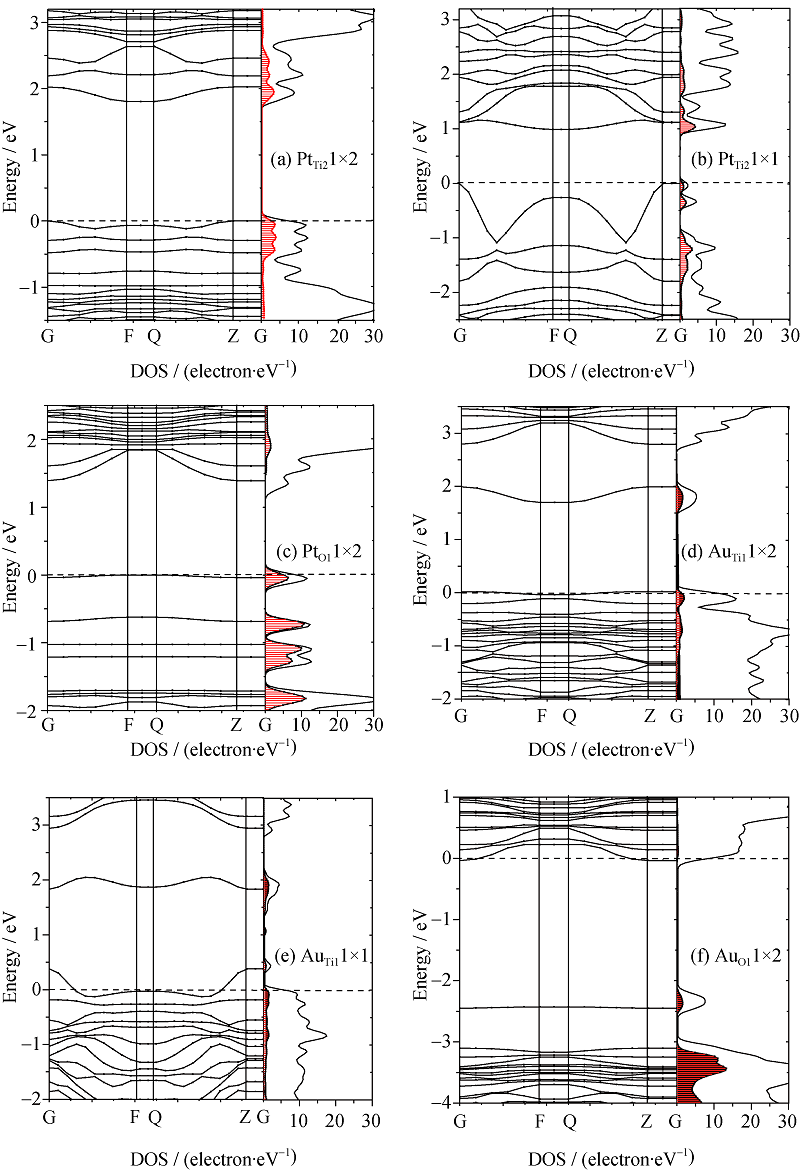 | 图3 贵金属修饰TiO2表面的能带结构和态密度图(填充部分为贵金属的分态密度)Fig. 3 The energy band structures and the density of states of TiO2(101) surface modified noble metal, whose shaded parts indicate the density of states of the impurity atoms and the standard dotted lines indicate the Fermi level |
前面分析已知, 富O条件更有利于实现Pt原子在Ti空位的较高浓度吸附。为此进一步计算了Pt原子吸附在Ti2空位位置的高浓度1× 1模型的电子结构。比较图3(a)和(b), 可知当杂质浓度提高时, 其价带顶上方的杂质能级变的很不平滑。在太阳光激发下, 该杂质能级上产生的空穴有效质量较小, 在外场的作用下加速度较大。与之相反, 导带底下方最低的杂质能级比较平坦, 其产生的电子有效质量较高, 在外场的作用下加速度较小, 因此高浓度的Pt掺杂有利于电子和空穴的分离, 提高量子产率。而且价带顶上方的最高表面能级为满填充状态, 它到导带底下方最低杂质能级的距离仅为1.12 eV, 显示出其对可见光有较强的吸收。图3(c)显示了Pt占据表面O1空位的位置时, Pt与周围Ti原子间的结合作用, 使Pt与周围等价六配位Ti的距离为0.236 nm, 与另两个不等价的五配位和六配位Ti的距离分别为0.232和0.251 nm, 这种Pt与周围Ti原子的非对称作用致使价带中出现劈裂峰, 增加了Pt 5d态的宽度[20], 以至于在价带顶附近出现了几条平坦的新杂质能级。
图3(d)显示了Au原子在Ti1空位位置的低浓度吸附的情形, 显示了在导带底下方0.70 eV附近出现了一条较深的表面能级, 这主要是表面悬挂键产生, 它容易成为光生电子和空穴的复合中心。此时Au与周围的O原子的杂化作用比较小, 且小于Pt在Ti2空位位置时与周围O原子的作用。当Au在表面在Ti1空位位置的掺杂浓度增加时, 显示了在导带底下方0.92 eV附近出现了一条较深的表面能级, 由表面悬挂键产生, 与低浓度吸附1× 2的情形相似。此时费米能级附近的杂质能级变的很不平坦, 说明了该杂质能级上的载流子有效质量较小, 有利于载流子的迁移, 如图3(e)所示。
图3(f)显示了当Au占据表面的O1空位的位置时, 在TiO2的价带顶上方约0.6 eV位置出现了一条表面能级, 这是由于Au的引入引起表面电荷分布改变引起的。而且其费米能级穿过导带底的下方, 其价带顶附近也有较多的Au杂质能级的分布, 使其表现出显著金属特征。事实上, 比较图3(c)和(f)可以看出, 还原条件下Pt对表面电子结构的影响大于Au, 这相似的结果在文献[21]讨论TiO2(110)面O空位时得到了解释。通过整体比较, 发现Au修饰的TiO2表面均显示出一定程度的金属性特征, 这与Pt修饰的具有半导体特征的表面能带结构有所不同。
Pt和Au原子吸附于化学计量比TiO2(101)面上或者间隙位置均显示有较高的正吸附能, 表明贵金属原子与化学计量比表面的吸附作用很弱, 因此对TiO2(101)面的电学性质影响较小。反之, 在富O条件下, 表面容易形成Ti空位, 此时Pt和Au原子吸附于该空位的位置时吸附能较低, 与Au原子不同, Pt原子有从TiO2表面扩散进入体相晶格中的趋势。而在富Ti条件下, 表面容易形成O空位, Pt和Au原子吸附于O空位的位置时吸附能较低。Pt和Au原子吸附在表面空位的位置, 可以“ 湿化” 该表面, 使其对表面的能带结构产生较大的影响, 突出表现在带隙中出现贵金属的5d杂质能级。另外, 在富O条件下制备出Pt修饰的表面为半导体特征, 而Au修饰的表面为金属特征。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|