



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TiO2/WO3微纳米纤维复合材料的制备及光催化性能
[孟姝虔 , 周德凤
, 周德凤
, 周德凤, 朱晓飞|
|


作者简介: 孟姝虔(1988-), 女, 硕士. E-mail:760632976@qq.com
通过溶胶-凝胶和静电纺丝技术相结合的方法, 成功制备不同复合浓度聚乙烯吡咯烷酮(PVP)/钛酸四正丁酯(Ti(OC4H9)4)/钨酸铵(N5H37W6O24·H2O)前驱体。通过控温煅烧获得不同煅烧温度、不同复合浓度的TiO2/WO3微纳米纤维复合材料。采用X射线衍射(XRD)、傅里叶变换红外光谱(FT-IR)、场发射扫描电子显微镜(FE-SEM)和紫外-可见漫反射光谱(UV-Vis )技术对样品进行表征。以亚甲基蓝(MB)的光降解为模型反应, 研究TiO2/WO3微纳米纤维复合材料在紫外光照射下的光催化活性。结果表明, 煅烧温度500℃时,
PVP/Ti(OC4H9)4/N5H37W6O24·H2O precursors with different Ti/W molar ratios were successfully synthesized by a simple combination method of Sol-Gel and electrospinning techniques. The electrospun precursors was converted into TiO2/WO3 micro-nanofibers
TiO2因化学性质稳定、无毒无害、价格低廉和催化效率高等优点而引起人们对其广泛应用的关注, 但其带隙宽(3.2 eV)、太阳光利用率不高、电子-空穴对复合率高导致其实际应用有限[ 1, 2, 3]。采用金属氧化物掺杂(V、W、Sr)[ 4, 5, 6, 7]、非金属氧化物掺杂(C、N、F)[ 8, 9, 10]、金属和非金属氧化物共掺杂[ 11, 12]、光催化剂光敏化[ 13]、半导体复合(Bi2WO6/TiO2[ 14]、V2O5/TiO2[ 15]、TiO2/ZnO[ 16]、TiO2/CdS[ 17])等方法可一定程度上提高TiO2的光催化性能。其中半导体复合改善光催化效果较为显著, 构成复合半导体光催化剂材料的导带和价带能级不同, 光生电子和空穴可通过运输而被分离, 减小光生电子和空穴的复合率, 从而提高了TiO2光催化活性[ 14, 15, 16, 17]。采用静电纺丝法制备的TiO2/ZnO复合纳米纤维, TiO2导带/价带的位置与ZnO导带/价带的位置交错排列, 更能有效地促进光生电子-空穴对的分离, 可实现复合纳米纤维对罗丹明B (RhB)的降解率接近100%, 对苯酚的降解率也能达到85%[ 16]。可见, 半导体复合法有可能控制光生电子和空穴的复合, 进而提高TiO2的光催化活性。
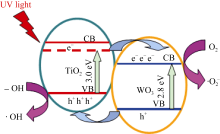
三氧化钨(WO3)作为窄带隙(2.8 eV) n型半导体, 因能够吸收可见光而成为理想的光催化剂[ 18, 19, 20]。WO3(频带边缘 ECB= 0.4 eV, EVB = 3.2 eV)[ 21]与TiO2( ECB= -0.5 eV, EVB = 2.7 eV)[ 22]耦合形成复合半导体光催化剂[ 23, 24],紫外光照射下TiO2和WO3同时发生带间跃迁, 由于导带和价带能级的差异, 光生电子聚集在WO3的导带上, 空穴聚集在TiO2的价带上, 使得光生电子和空穴得到有效分离, 因而TiO2的光催化活性得到提高。
不同的制备方法影响材料的结构、形态和性能[ 25, 26, 27, 28]。溶胶-凝胶法和控温煅烧法制备的WO3/TiO2复合纳米棒[ 25], 在煅烧温度800℃、WO3的复合浓度为4wt%时, 紫外光照射下对罗丹明B(RhB)的光催化效率可达95%; 超声喷雾热分解法制备的TiO2/WO3多孔微球[ 26], 当 n(W)/ n(Ti)为0.04时太阳光照射40 min对乙醛的降解效率比TiO2提高1.56倍。因此, 本研究采用静电纺丝法和控温煅烧法制备TiO2/WO3微纳米纤维复合材料, 通过光催化降解亚甲基蓝(MB)对其光催化活性进行评价, 讨论煅烧温度和复合比例对TiO2/WO3复合微纳米纤维结构和光催化性的影响, 分析和探讨TiO2/WO3复合微纳米纤维的光催化机理。
所用钨酸铵(N5H37W6O24·H2O)、钛酸四正丁酯(Ti(OC4H9)4)、聚乙烯吡咯烷酮(PVP,[Mn] = 1300000)均为分析纯试剂(Alfa Aesar); 无水乙醇(CH3CH2OH)、冰醋酸(CH3CH2COOH)均为分析纯试剂(北京化学试剂公司)。
称取1.4 g PVP, 将其加入装有10 mL无水乙醇的锥形瓶中, 封口放在磁力搅拌器上搅拌约10 h得PVP溶液。另外量取1 mL钛酸四正丁酯(Ti(OC4H9)4)溶液溶解到2 mL乙酸和2 mL无水乙醇中, 搅拌约12 h。称取0.3925 g钨酸铵溶解在4 mL无水乙醇中将其封口搅拌2 h。然后将上述两种混合液( n(Ti): n(W) = 2: 1)缓慢加入到PVP溶液中, 继续搅拌12 h即得到粘稠PVP/Ti(OC4H9)4/N5H37W6O24·H2O溶胶。实验过程中用塑料薄膜对容器封口。
将上述配置好的前驱体溶胶倒入注射器中, 并将金属电极插入注射器内。调节注射器倾斜角度使其大约与水平面成30°,调整毛细管尖端与铝薄接收板的距离约为16 cm, 施加16 kV电压。随着接收时间的增加, 最终在铝薄接收板上得到纤维毡。纺丝过程中每隔一段时间需用干净的滤纸擦拭毛细管管口。纺丝结束后用尖镊子撕下制备的纤维毡真空干燥。将干燥后的纤维毡剪成适当大小置于坩埚内, 于马弗炉中以1 ℃/min速度升温并分别在400、500、600、700 和800℃时保持温度3 h, 自然降至室温得到所需样品。
在相同条件下, 改变 n (Ti): n(W)的比例为12:1、10:1、8:1、6:1、4:1和2:1, 在500℃下保温3 h后自然降至室温得所需样品, 分别命名为TW-12、TW-10、TW-8、TW-6、TW-4、TW-2。相同实验条件下静电纺丝制备纯TiO2和WO3样品。
采用日本理学D/Max-IIB型X射线衍射仪检测样品, Cu Kα1为射线源(λ = 0.15405 nm), 工作电压40 kV, 工作电流20 mA, 扫描速度4°/min, 扫描范围2 θ = 10°~80°; 红外光谱分析采用美国Nicolate公司的670型FT-IR, KBr压片, 扫描范围为500~4000 cm-1; 采用日本理学FEI-Philips XL-30 场发射扫描电子显微镜(Field Emission Scanning Electron Microscopy, FE- SEM)观测样品形貌; 采用美国Perkin Elmer公司的Lambda 900 UV-Vis-NIR分光光度计测试样品的紫外-可见漫反射光谱, BaSO4压片; BET比表面积测试用Micromeritics公司的全自动程序升温物理化学吸附仪AutoChem II 2920测定; 样品吸光度采用UV-Vis 756 B分光光度计测试。静电纺丝和光催化设备为自组装的装置。
取10 mg/L的MB水溶液100 mL置于高20 cm、体积为250 mL的玻璃空心柱中, 分别加入0.05 g制备的TiO2/WO3微纳米纤维复合材料, 在无光照条件下搅拌30 min使其达到吸附-脱附平衡。然后置于自组装的光催化反应器中, 反应器安装冷却水循环系统维持反应体系处于室温, 采用50 W高压汞灯作为紫外光源(发射波长365 nm), 照射间距为30 cm。磁力搅拌后分别在20、40﹑60﹑80﹑100、120和140 min取样, 将所得溶液离心分离后弃上清, 用UV-Vis分光光度仪测定溶液在波长为664 nm处吸光度的变化。根据吸光度的变化判定材料对MB的降解效果。降解效果以脱色率 D%表示:
D= [( A0- A)/ A0] ×100 % (1)
A0: 染料溶液最大吸收峰的初始吸光度; A: 染料溶液最大吸收峰的最终吸光度。
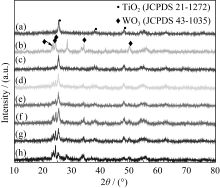
不同热处理温度下样品的XRD测试结果如图1所示。图1(a)为纯TiO2经不同温度煅烧的结果。煅烧温度400℃时位于25.2°、37.8°和48.0°的衍射峰分别归属于锐钛矿TiO2(JCPDS 21-1272)的(101)、(004)和(200) 晶面; 随着煅烧温度上升至500℃, TiO2特征峰更加尖锐; 煅烧温度为600和700℃时位于27.3°和36.0°处的新衍射峰归属于金红石相TiO2(JCPD-S21-1276)的(110)和(101)晶面, 说明600℃时TiO2的晶型由锐钛矿相向金红石相发生转变, 600和700℃时TiO2的锐钛矿和金红石相同时存在; 煅烧温度为800℃时, TiO2的锐钛矿完全转变为金红石相。
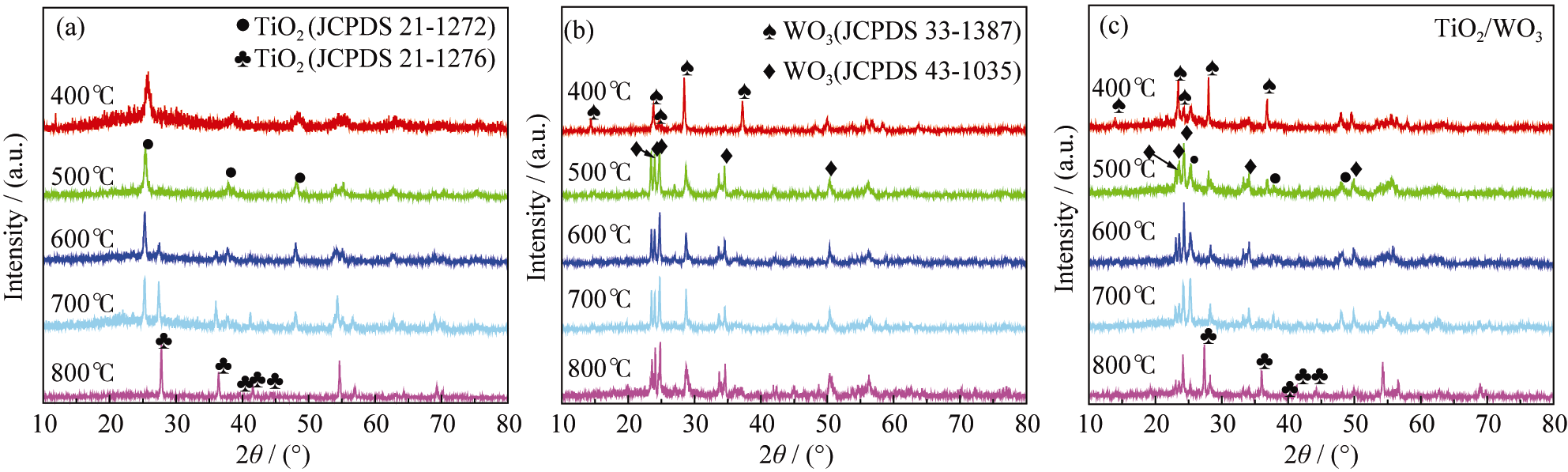 | 图1 纯TiO2 (a)、纯WO3 (b)和TiO2/WO3(c)样品不同煅烧温度的XRD图谱Fig. 1 XRD patterns of pure TiO2(a), pure WO3 (b) and TiO2/ WO3 composite (c) samples calcined at different temperatures |
图1(b)为纯WO3经不同温度煅烧的结果。煅烧温度在400℃时位于13.9°、22.64°、24.36°、28.2°和36.5°处的衍射峰, 归属于六方晶系WO3(JCPDS 33-1387)的(110)、(001)、(110)、(200)和(201)晶面; 煅烧温度升高至500℃时, 六方晶系WO3的部分衍射峰消失, 在23.1°、23.6°、24.4°、34.2°和50°出现新的衍射峰, 分别归属于单斜晶系WO3(JCPDS43- 1035)的(002)、(020)、(200)、(202)和(140) 晶面; 煅烧温度上升到600、700、800℃时, 各个衍射峰强度逐渐增强, 峰宽逐渐变窄变尖, 表明单斜晶系WO3结晶度提高。
以复合浓度 n(Ti): n(W) = 2:1为例,考察煅烧温度对TiO2/WO3复合物晶型的影响, 结果如图1(c)所示。煅烧温度400℃时呈现六方晶系WO3的衍射峰和微弱的锐钛矿TiO2衍射峰, 说明WO3的加入可抑制锐钛矿TiO2的形成; 煅烧温度500℃时六方晶系WO3的衍射峰部分消失, 单斜晶系WO3的衍射峰出现, 25.2°、37.8°和48.0°出现的衍射峰与锐钛矿TiO2的衍射峰完全吻合, 说明温度从400℃上升到500℃时WO3由六方晶系转变为单斜晶系, 同时锐钛矿TiO2形成; 热处理温度上升至600℃、700℃时各个衍射峰都细锐化, 证实TiO2/WO3复合物晶型逐渐完善; 煅烧温度800℃时锐钛矿TiO2的衍射峰消失, 金红石相TiO2出现, 说明800℃时TiO2的晶型完全由锐钛矿相转变为金红石相。可见, 纯TiO2由锐钛矿相转变为金红石相的转变温度是600℃, TiO2/WO3复合后晶相的转变温度升高至800℃, 说明WO3的复合改变了TiO2的相转变温度。不同晶型的TiO2其催化性能不同[ 29], 因此WO3的加入将对研究体系的光催化性能产生一定的影响。
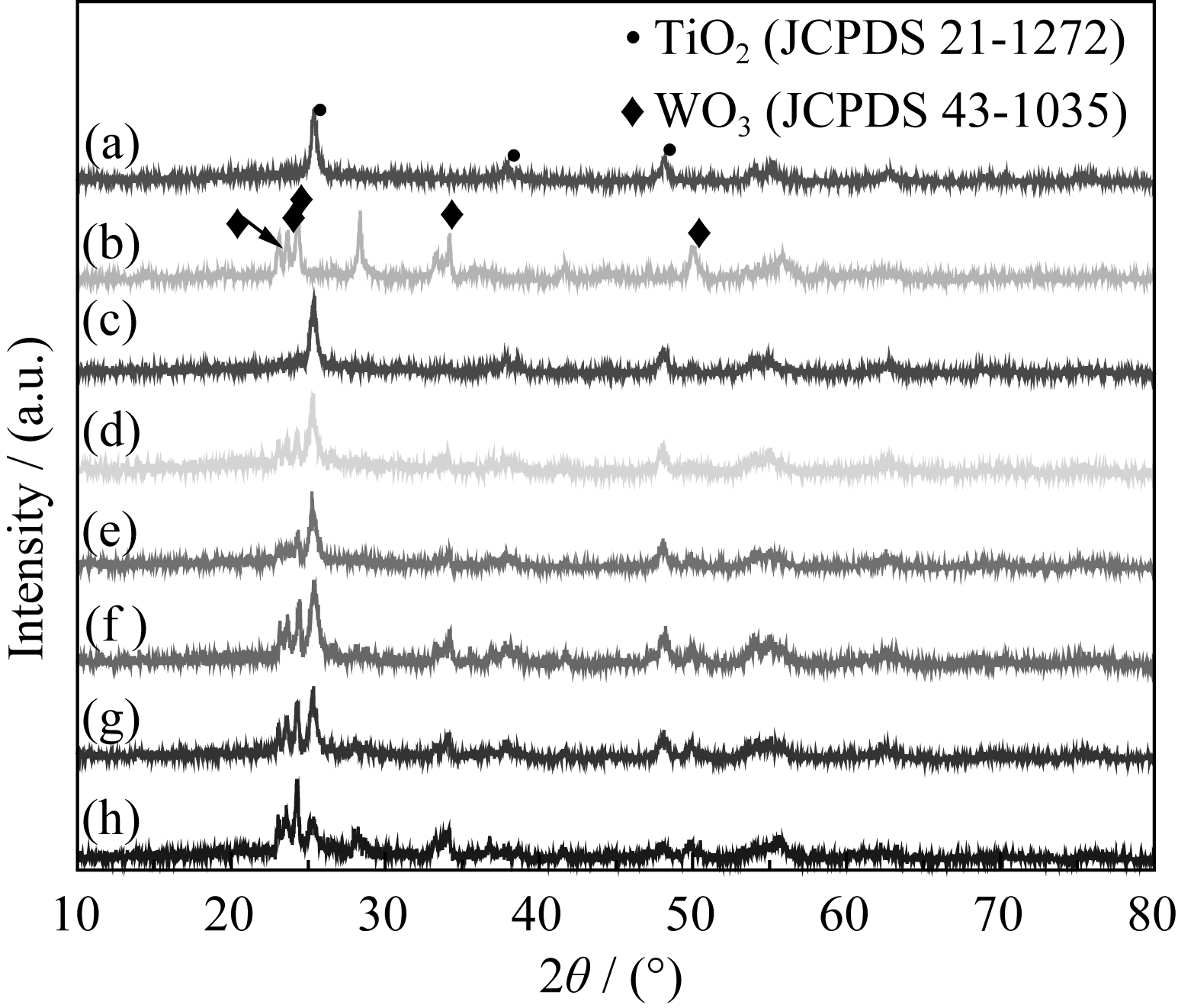 | 图2 煅烧温度500℃时TiO2、WO3和不同复合浓度的TiO2/WO3微纳米纤维的XRD图谱Fig. 2 XRD patterns of TiO2 ,WO3 and TiO2/WO3 micro- na-no-f-ibers with different Ti/W molar ratios calcined at 500℃(a) TiO2; (b) WO3; (c) TW-12; (d) TW-10; (e)TW-8; (f) TW-6; (g)TW-4; (h) TW-2 |
为考察复合浓度n(Ti):n(W)对TiO2/WO3结构的影响, 图2给出500℃煅烧样品的XRD图谱。当n(Ti):n(W) = 12:1时(图2(c)), 所有的衍射峰都归属于锐钛矿相TiO2, 没有WO3以及其它复合物出现, 说明掺杂的W6+完全进入TiO2晶格。随着WO3复合浓度增加, 即n(Ti):n(W)比由10:1变化至2:1, 图2(d)~(h)中TiO2/WO3复合结构中锐钛矿相TiO2的衍射峰明显宽化, 单斜晶系WO3的衍射峰逐渐增强, 没有金红石相TiO2出现。由此可见, n(Ti):n(W) = 12:1时掺杂的W6+完全进入TiO2晶格, n(Ti):n(W) = 10:1、8:1、6:1、4:1、2:1时, 掺杂的W6+除部分进入TiO2 晶格外, 剩余的WO3与TiO2形成TiO2/WO3复合氧化物。
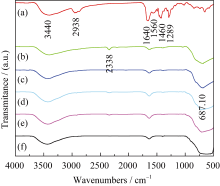
以 n(Ti): n(W) = 2:1为例, 对前驱体纤维PVP/ Ti(OC4H9)4/N5H37W6O24·H2O及不同温度处理的TiO2/WO3复合微纳米纤维进行FT-IR测试, 结果如图3所示。由图可以看出, 3440 cm-1与1640 cm-1处的吸收峰分别对应吸附水和羟基的伸缩振动, 表面羟基对O2的吸附能力较强, 可以提高TiO2光催化活性[ 30]; 热处理前(图3(a))在2938、1560、1460和1289 cm-1处的吸收峰分别归属于C-H、C-C、C-O和O-H的伸缩振动峰[ 31, 32]; 煅烧温度400℃(图3(b))时有机吸收峰明显减弱或消失, 表明经过煅烧后有机物脱除, 2338 cm-1处出现新的吸收峰归属于CO2的特征峰[ 30]; 煅烧温度500℃时(图3(c))归属于PVP有机物的振动峰消失,687 cm-1处出现新的振动峰归属于Ti-O的伸缩振动,同时在700~900 cm-1 (766 cm-1, 815 cm-1) 处出现W-O振动峰[ 31, 32]; 煅烧温度600、700和800℃时(图3d~f)没有出现新的吸收峰, 687 cm-1处和700~900 cm-1处的振动峰有所加强。由此可得, 煅烧温度500℃以上, PVP/Ti(OC4H9)4/N5H37W6O24·H2O的有机成分完全分解, 形成TiO2/WO3复合氧化物。这与图1(c)的XRD结果一致。
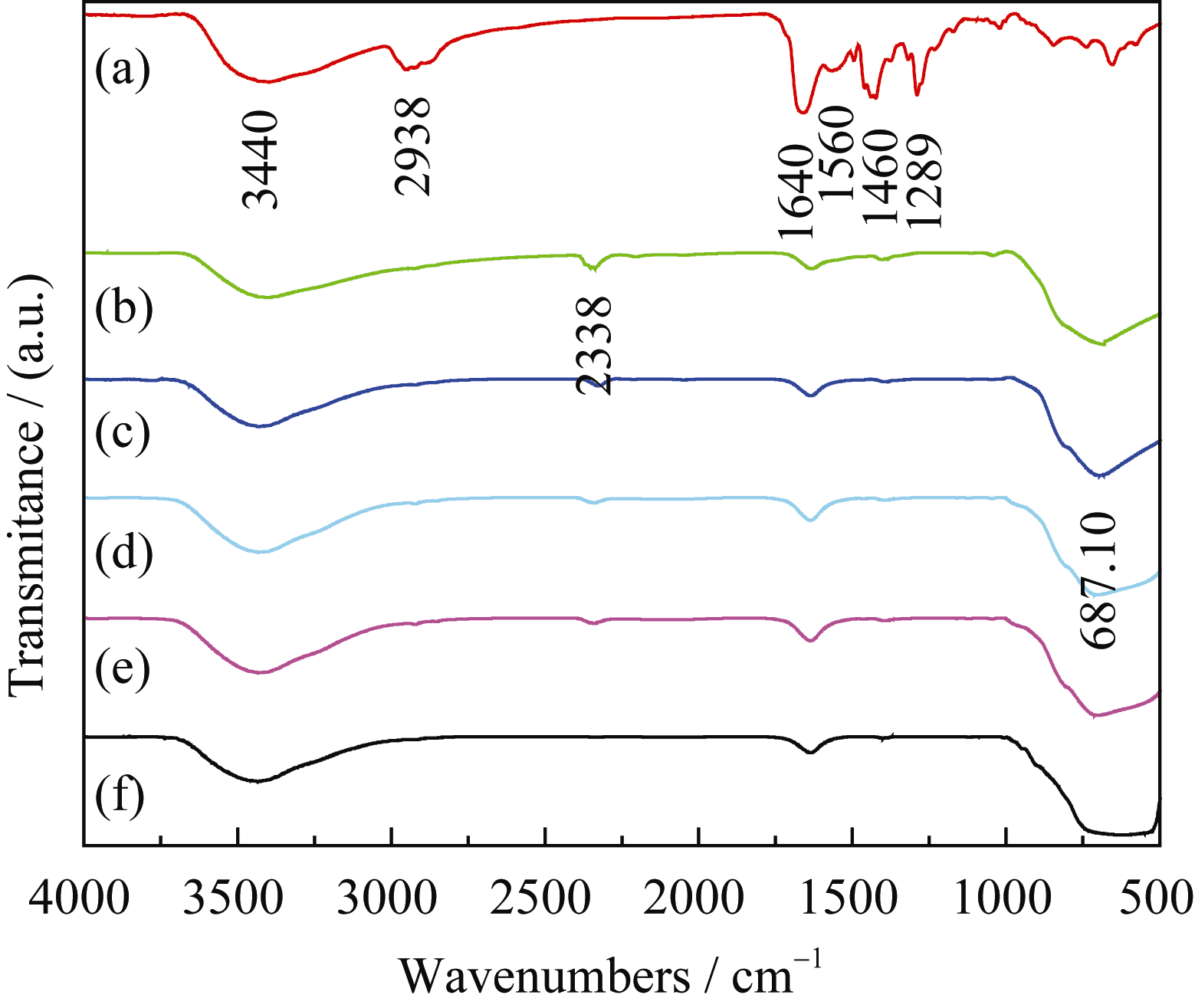 | 图3 PVP/Ti(OC4H9)4/N5H37W6O24·H2O( n(Ti): n(W) = 2:1)纤维热处理前后的红外光谱图Fig. 3 FT-IR spectra of the precursors calcined at different temperatures(a) PVP/Ti(OC4H9)4/N5H37W6O24·H2O ( n(Ti): n(W)=2:1) precursors, (b)-(f) Precursors calcined at 400, 500, 600, 700 and 800℃, respectively |
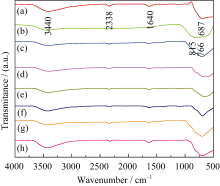
煅烧温度达到500℃时前驱体中的有机成分完全分解(图3), 因此以500℃煅烧样品为例, 测试不同复合浓度 n(Ti): n(W)样品的红外光谱, 结果如图4所示。687 cm-1处的吸收峰归属于TiO2的Ti-O伸缩振动; 图4(b)中700~900 cm-1(766 cm-1, 815 cm-1)处的吸收峰归属于W-O的伸缩振动; 当 n(Ti): n(W) = 12:1时(图4(c)), 在600~900 cm-1范围内只出现Ti-O振动峰, 没有W-O振动峰, 说明掺杂少量W6+没有破坏TiO2晶格; 图4(d)~(h)与图4(a)、4(b)比较可知, 在600~900 cm-1范围内均出现Ti-O和W-O的振动峰, 说明复合浓度 n(Ti): n(W)为10:1、8:1、6:1、4:1和2:1时, 形成TiO2/WO3复合氧化物, 与上述XRD (图2)的结果一致。
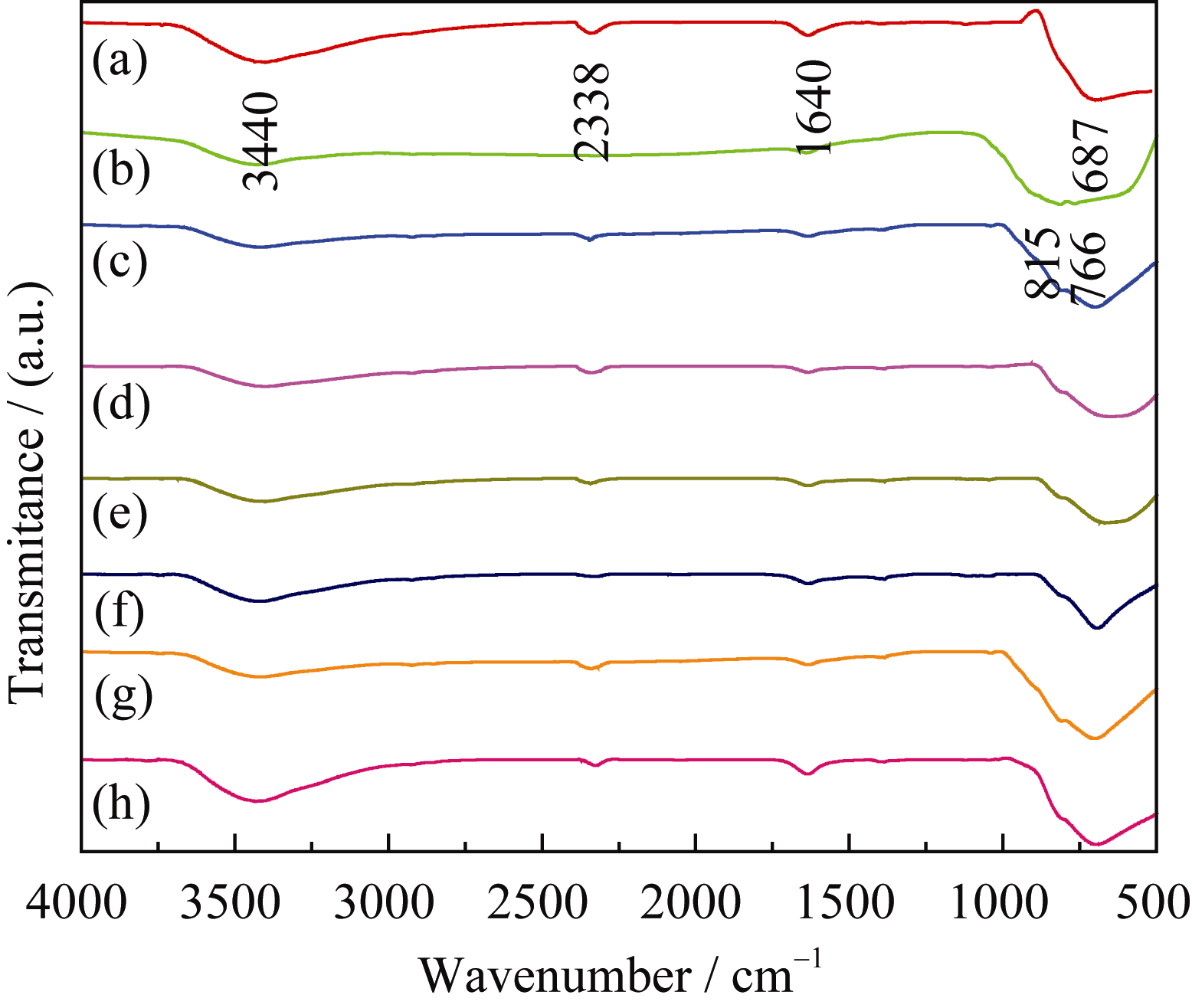 | 图4 煅烧温度500℃时TiO2、WO3和不同复合比例的TiO2/WO3微纳米纤维的红外图谱Fig. 4 FT-IR patterns of TiO2, WO3 and TiO2/WO3 micro- nan-o-fiber composites with different Ti/W molar ratios calcined at 500℃(a) TiO2;(b) WO3;(c) TW-12; (d) TW-10; (e) TW-8; (f) TW-6; (g) TW-4; (h) TW-2 |
 | 图5 500℃煅烧的TiO2、WO3和不同复合浓度的TiO2/WO3微纳米纤维的FE-SEM照片Fig. 5 SEM images of TiO2, WO3 and TiO2/WO3 micro-nanofibers with different Ti/W molar ratios calcined at 500℃(a) TiO2; (b) WO3; (c) TW-8; (d) TW-6; (e) TW-4; (f ) TW-2; (g) Magnified FE-SEM image of TW-4; (h) Magnified FE-SEM image of TW-2 |
图5为500℃热处理样品的FE-SEM照片。图5(a)为表面光滑、尺寸分布均一、纤维间没有交联的TiO2微纳米纤维, 直径200 nm; 图5(b)为表面光滑且结晶度良好的WO3微纳米粒子; 图5(c)~(f)为不同复合浓度的TiO2/WO3微纳米纤维的FE-SEM照片。当 n(Ti): n(W) = 8:1和6:1 时(图5(c)~(d)), TiO2/WO3微纳米纤维仍保持细直、光滑均匀的形貌; 随着WO3复合浓度进一步增大至 n(Ti): n(W) = 4:1和2:1时(图5(e)~(f)), 细直的纤维出现断裂, 复合纤维的长/经比明显减小。这是因为, 500℃煅烧时, 有机物和钨酸铵分解形成气体[ 20, 32](NH3、H2O、NO、CO2、N2O), 随着钨酸铵复合浓度的提高, 产生大量的气体冲破表层纤维, 纤维在热应力作用下被撕裂, 所以导致纤维断裂; 另外, 当钨酸铵的浓度增大到一定值(TW-2)时, 静电纺丝形成的复合微纳米纤维的稳定性增强[ 20], 导致样品TW-2的断裂程度不如样品TW-4。由样品TW-4、TW-2放大的FE-SEM照片(图5(g)~(h))可见, TiO2/WO3复合微纳米纤维的表面不再光滑, 少量的WO3微纳米粒子负载于TiO2微纳米纤维上, 并且随着WO3复合浓度的提高, 负载于断裂TiO2微纳米纤维表面的WO3微纳米粒子数目增多, 这与XRD(图2)表征的结果一致。
| 表1 500℃煅烧的TiO2、WO3和不同复合浓度的TiO2/WO3微纳米纤维的比表面积 Table 1 Specific surface area of TiO2, WO3 and TiO2/WO3 micro-nanofibers with different Ti/W molar ratios calcined at 500℃ |
由各样品的比表面积(表1)可以看出:样品TW-4(40.97 m2/g)和TW-2(17.92 m2/g)的比表面积大于TiO2微纳米纤维(8.48 m2/g), 复合样品比表面积的增大归因于WO3微纳米颗粒的引入; 然而, 当WO3的复合量超过一定限度时(TW-2), 过多的WO3颗粒可阻塞复合微纳米纤维的部分孔隙, 从而导致TW-2比表面积减小[ 26]。催化剂的比表面积是影响其光催化活性的重要因素。TW-4具有较高的光催化活性, 是由于其较大的比表面积可以大量地吸附溶液中的有机物和强氧化剂 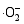
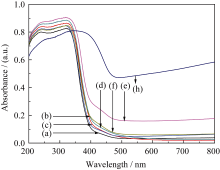
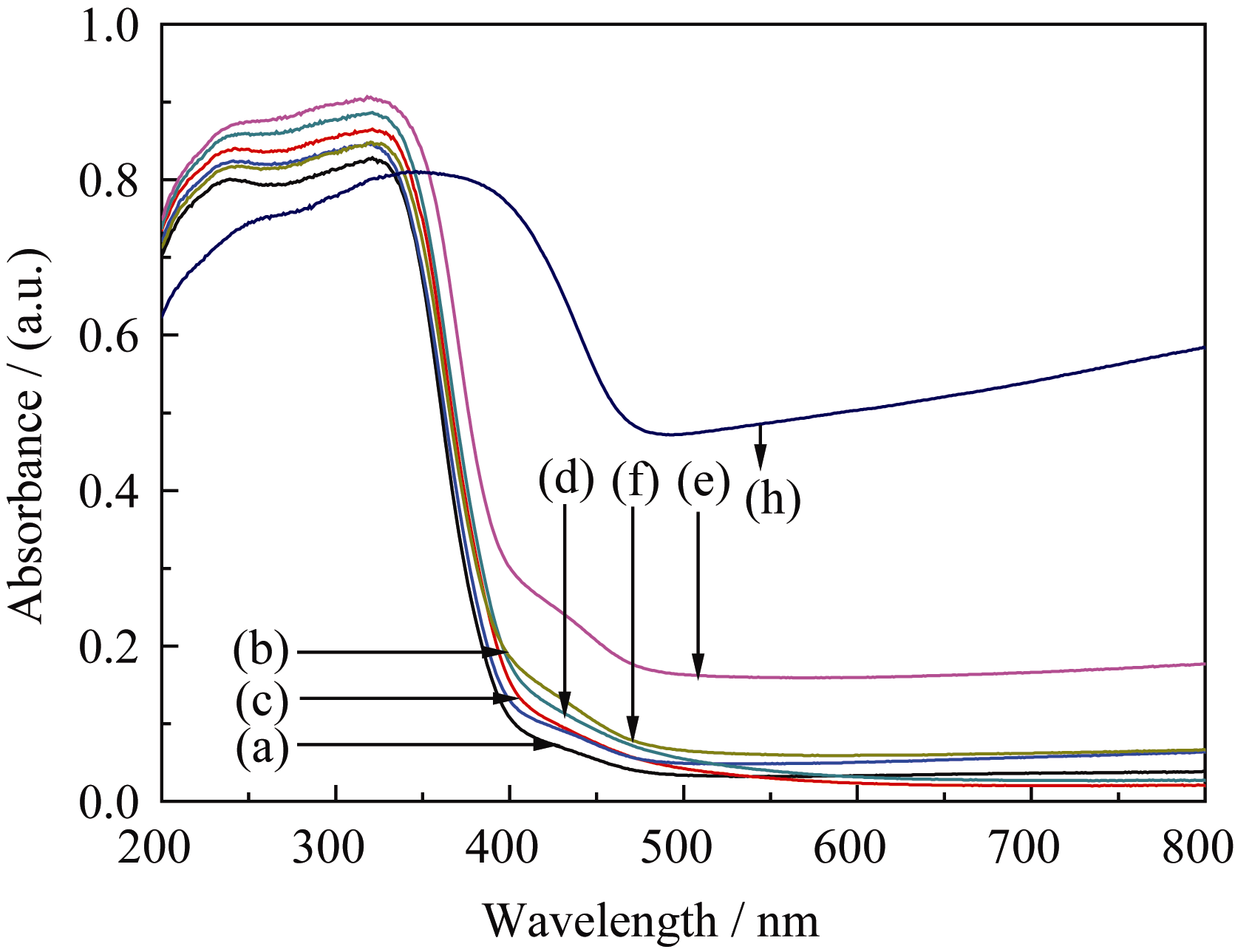 | 图6 500℃煅烧的TiO2、WO3和不同复合浓度的微纳米纤维的UV-Vis漫反射光图谱Fig. 6 UV-Vis diffuse reflectance spectra of TiO2, WO3 and TiO2/WO3micro-nanofibers with different Ti/W molar ratios calcined at 500℃(a) TiO2; (b) TW-12; (c) TW-8; (d)TW-6; (e) TW-4; (f ) TW-2; (h) WO3 |
为研究合成样品的光吸收性能, 对500℃煅烧的不同复合浓度TiO2/WO3微纳米纤维进行UV-Vis-NIR/DR测试, 结果如图6所示。从图6(a)可知, 纯TiO2的吸收带边约为390 nm; 在图6(c)~(f)中发现存在两个明显的吸收边界, 这主要归因于TiO2和WO3的特征吸收[ 12, 16, 33], 说明TiO2/WO3微纳米纤维复合材料由TiO2和WO3组成, 这与XRD和FT-IR的结果相一致。随着WO3含量的增加, TiO2/WO3复合氧化物的吸收带边发生不同程度的红移, TW-12、TW-8、TW-6、TW-4、TW-2 吸收边带分别约为425、410、440、480、450 nm。然而, 图中吸收峰的红移与图6中复合材料500℃煅烧的TiO2、WO3和不同复合浓度的微纳米纤维的UV-Vis漫反射光谱图中WO3含量的增加并不一致, WO3掺杂于TiO2形成的TW-12比纯TiO2吸收边带增加35 nm; 复合材料中TW-4的光吸收最强, 比纯TiO2吸收边带增加90 nm。TiO2/WO3微纳米纤维在紫外区呈现较强吸收, 可能是W6+离子掺入TiO2引起晶格缺陷, 改变了TiO2的能级结构; 也可能是TiO2与WO3形成复合氧化物, 促进光生电子-空穴对的分离; 亦或是两种情况共同作用的结果。禁带宽度 Eg(eV)与吸收极限 λ0(nm)的换算公式如下:
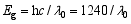
式中h = 6.62176×10-34J·s,为普朗克常数; c =3×108m/s,光速; 吸收极限 λ0由吸收边做出切线与横轴的交点确定。根据公式(2)推算出TiO2、TW-12、TW-8、TW-6、TW-4和TW-2的带隙宽度分别为3.18、2.92、3.02、2.82、2.58和2.76 eV。由此可知, 不同复合浓度的复合氧化物光吸收边带红移越多, 拥有越窄的带隙能; 带隙能的大小影响样品的光催化性能, 提示TW-12的光催化活性高于TiO2, 复合样品中TW-4可能是最有效的光催化剂。
2.5.1 不同煅烧温度TiO2/WO3微纳米纤维复合材料的光催化性能
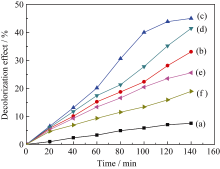
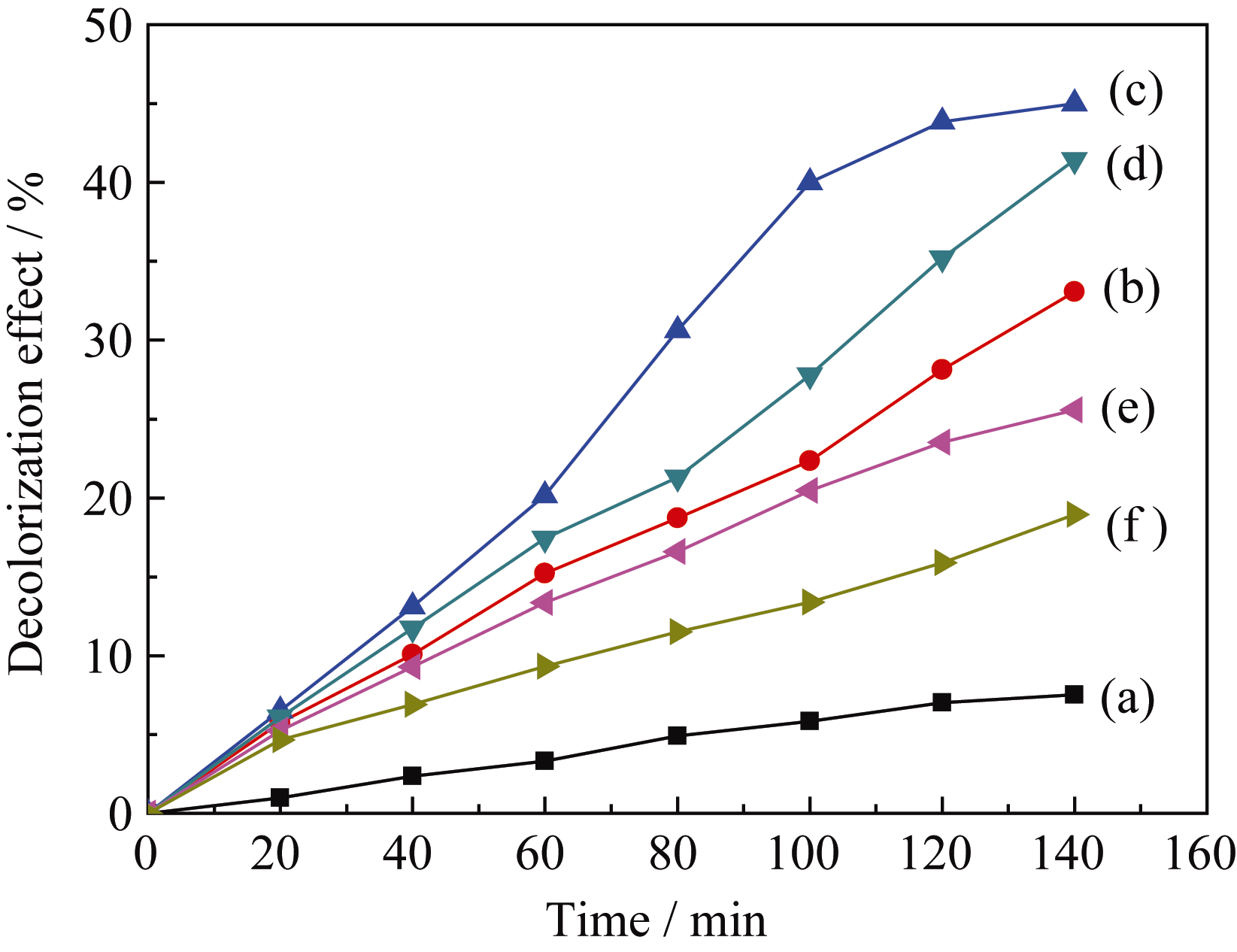 | 图7 TiO2/WO3复合微纳米纤维(n(Ti): n(W) = 2:1)在不同煅烧温度下对MB的光降解曲线Fig. 7Fig. 7 Photocatalytic degradation of MB over TiO2/WO3 (n(Ti): n(W) = 2:1) micro-nanofibers composites calcined at different temperatures under UV light irradiation (a) MB; (b) Calcined at 400℃; (c) Calcined at 500℃; (d) Calcined at 600℃; (e) Calcined at 700℃; (f) Calcined at 800℃ |
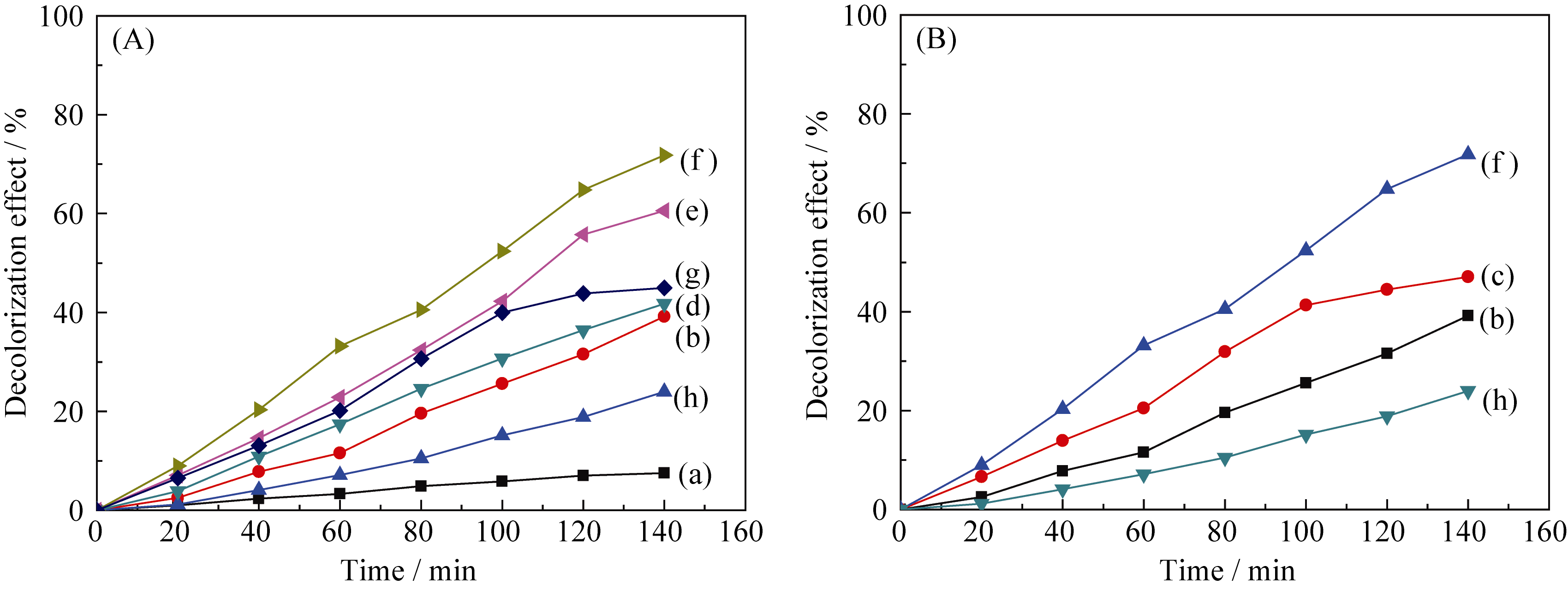 | 图8 TiO2、WO3和不同复合浓度的TiO2/WO3微纳米纤维对MB的光降解曲线Fig. 8 Photocatalytic degradation of MB over TiO2, WO3 and TiO2/WO3 micro-nanofibers with different Ti/W molar ratios under UV light irradiation (a) MB; (b) TiO2; (c) TW-12; (d) TW-8; (e)TW-6; (f ) TW-4; (g) TW-2; (h) WO3 |
以MB为光降解模型探讨不同煅烧温度对TiO2/WO3( n(Ti): n(W) = 2:1)微纳米纤维复合材料在紫外光照射下的光催化性能, 结果如图7所示。由图7可知, 不同温度煅烧的样品均表现一定的光催化活性, 且随着光照时间的增加对MB的降解率增加。比较图7(b)~(f)可知随着煅烧温度由400℃上升至800℃, TiO2/WO3微纳米纤维复合材料对MB的光降解效率先升高后下降, 140 min后, 光降解效率依次为33.08%、45.0%、41.44%、25.58%、18.95%; 其中500℃煅烧的TiO2/WO3微纳米纤维对MB的催化活性最好。这是因为煅烧温度400℃时, 催化活性较好的锐钛矿TiO2没有形成, 只生成六方晶系WO3; 煅烧温度500℃时, 锐钛矿TiO2和单斜晶系WO3生成, 单斜晶系WO3的光催化活性高于六方晶系WO3[ 19], 因而500℃煅烧的TiO2/WO3复合微纳米材料的催化活性高于400℃; 煅烧温度升高至600、700℃, 锐钛矿TiO2和单斜晶系WO3晶体的结晶度提高, 晶粒尺寸变大, 表面积减小, 从而降低了光催化活性; 煅烧温度进一步升高到800℃时, TiO2由锐钛矿相转变为金红石相, 因而复合氧化物的光催化活性降低。另外, 500℃煅烧的样品其锐钛矿TiO2结构不完善, 存在很多缺陷, 可以吸附更多的 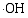
为比较WO3掺杂TiO2和TiO2与WO3复合形成的TiO2/WO3微纳米纤维两种样品的光催化性能,对掺杂样品TW-12和复合样品TW-4的光催化性能进行比较, 结果如图8(B)所示。经过140 min的光催化反应, 样品TW-12和TW-4对MB的光催化效率高于纯TiO2, 分别为47.06%和71.82%。由表1比表面积数据可知, 样品TiO2(8.48 m2/g)和TW-12(9.04 m2/g))的比表面积相差不大, 因此比表面积的增加对TW-12光催化活性的影响不是主要因素。半导体的光催化活性很大程度上取决于其禁带宽度的大小, 禁带宽度越窄, 可见光的利用率就越高, 光催化活性就越高[ 16]。由UV-Vis-NIR/DR分析可知, TiO2、TW-12和TW-4吸收边带分别在390、425和480 nm, 对光的利用率大小依次为TW-4 > TW-12 >TiO2。样品TW-4具有较高的光催化活性, 是其大的比表面积(40.97 m2/g)、W6+进入TiO2晶格形成的晶格缺陷和TiO2与WO3间形成复合氧化物等综合作用的结果。
2.5.3 光催化机理
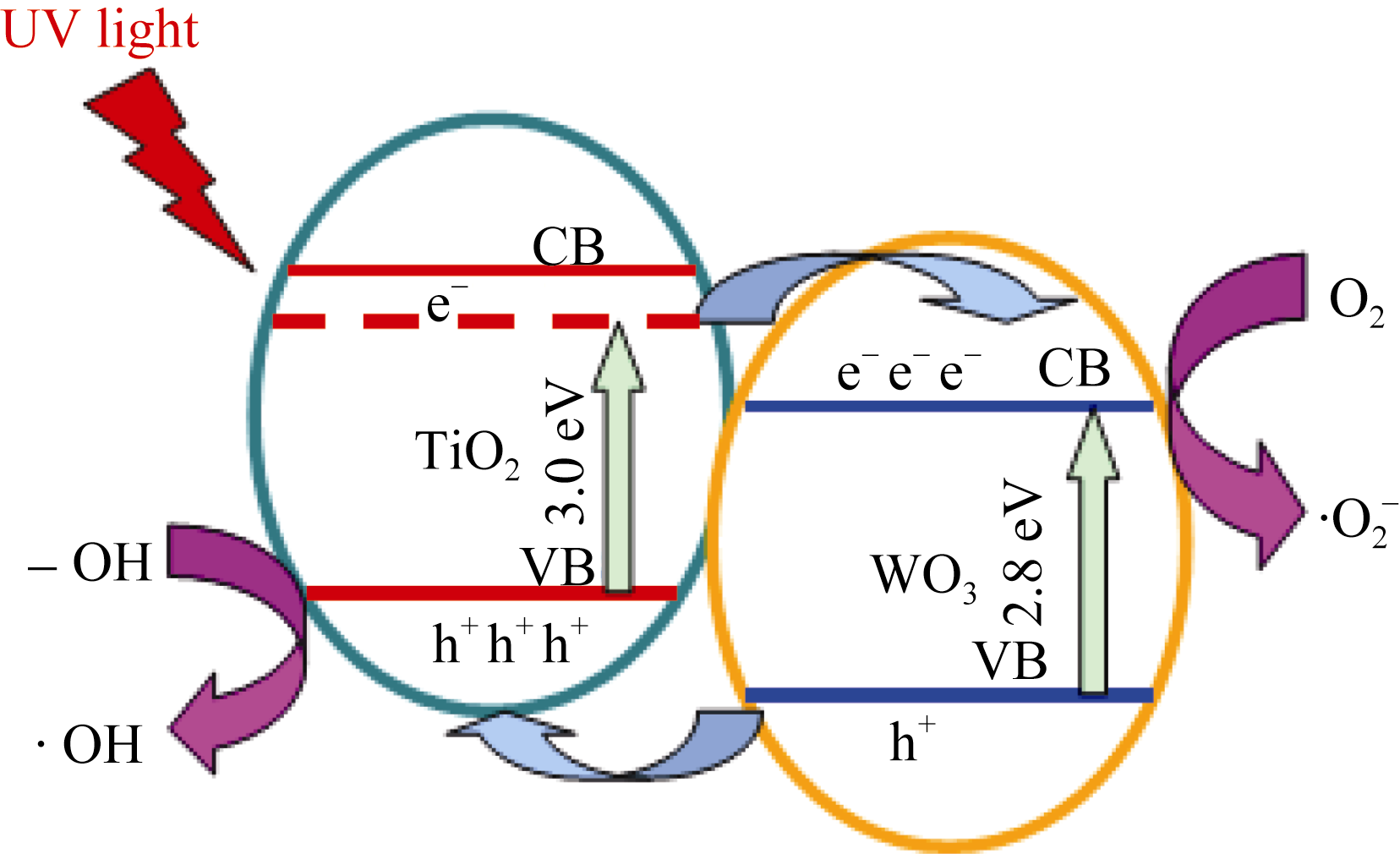 | 图9 TiO2/WO3复合微纳米纤维的催化机制示意图Fig. 9 Mechanism schematic diagram of TiO2/WO3 micro- nanofiber composites |
由光催化实验数据可知, 无论是WO3掺杂TiO2所形成的TW-12, 还是不同浓度WO3复合形成的TiO2/WO3微纳米纤维的光催化活性高于均纯TiO2。主要原因如下: 其一, 由于掺杂的WO3和 TiO2对应的半导体带隙不同, 当部分W6+置换部分Ti4+离子, 形成的缺陷方程式如下:
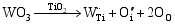
在体系中会形成晶格缺陷, 从而导致体系带隙的变化; 由于禁带值变小, 电子激发时跃迁距离缩短, 电子空穴对分离效率提高, 致使光催化反应能产生大量的 
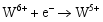
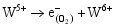
其二, TiO2与WO3之间形成复合氧化物, TiO2/WO3微纳米纤维的光催化机制如图9所示。当有恰当的光源照射在TiO2/WO3复合半导体光催化剂上时, TiO2和WO3价带上的电子被激发, 由于TiO2中掺杂能级的存在使得TiO2的导带位置下降, 这些掺杂能级使得TiO2价带中电子吸收较小能量的光子就能跃迁到TiO2的导带上, 由于TiO2和WO3的导带之间存在着能级差异, WO3的导带能级较低, 有利于TiO2导带上的光生电子转移到WO3的导带并聚集; 同理, 在TiO2的价带上形成空穴的聚集, 这样使得光生电子和空穴有效地分离, 导致复合微纳米纤维的光催化活性的提高。其具体催化过程如下[ 17, 22, 24, 26]:
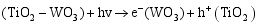
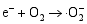
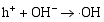
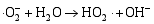
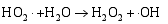
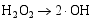
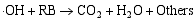
在催化剂的表面光生空穴被 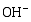
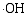
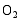
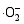
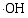
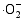
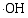
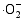
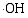
采用溶胶-凝胶与静电纺丝相结合方法制备了PVP/Ti(OC4H9)4/N5H37W6O24·H2O前驱体, 通过控温煅烧获得TiO2/WO3复合微纳米纤维。WO3微纳米颗粒的引入使TiO2的相变温度从600℃上升到800℃, 复合样品比表面积增大。WO3掺入TiO2晶体形成掺杂能级, 以及半导体TiO2和WO3之间的能级差, 有效促进光生电子-空穴对的分离, 提高光催化活性。无论是WO3掺杂TiO2形成的TW-12样品, 还是WO3与TiO2形成的复合氧化物TW-4, 在紫外光照射下对MB的光降解效率均高于纯TiO2, TW-4对MB光降解效率可高达71.82%。TiO2和WO3复合材料是一种有潜力的光催化剂。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|