
{kind=link}
{kind=link}
{kind=link}
{kind=link}
铋层状共生结构铁电体Bi7Ti4NbO21的第一性原理计算
[刘峰1, 2  , 陆毅青
, 陆毅青1 , 李永祥1 ]
, 陆毅青]
|
|


刘 峰(1987-), 男, 博士研究生. E-mail:liufeng@student.sic.ac.cn
铋层状共生结构铁电体Bi7Ti4NbO21具有天然的超晶格结构。采用基于密度泛函(DFT)的第一性原理计算对Bi7Ti4NbO21及其相关化合物进行了研究。计算得到Bi7Ti4NbO21的生成焓为-56.21 eV, 带隙为0.796 eV, 而对于组成单元Bi3TiNbO9和Bi4Ti3O12, 生成焓分别为-30.72和-43.32 eV, 带隙分别为2.535和2.436 eV。分析表明, 共生结构Bi7Ti4NbO21相对于同组分的组成单元处于热力学亚稳态, 其钙钛矿层是电子电导的主要影响因素, 带隙相对于组成单元减小也是源于钙钛矿层的导带底的下移。
Intergrowth bismuth layer-structured ferroelectric Bi7Ti4NbO21 material (
Aurivillius1950年首先发现了铋层状结构铁电材料[ 1]。该类材料的化学通式为(Bi2O2)2+(A m-1B mO3 m+1)2-, m为每两个相邻(Bi2O2)2+层之间包含的氧八面体的层数。不同层数 m的铋层状化合物可以沿 c轴交替共生, 形成一种超晶格结构, 这种结构称为铋层状共生结构化合物。例如, 由Bi3TiNbO9 ( m=2, BTN)和Bi4Ti3O12( m=3, BiT), 沿 c轴交替共生形成Bi7Ti4NbO21 ( iBTN)。铋层状结构材料不仅具有居里温度高、介电常数低、老化率低、介电击穿强度高、谐振频率的时间和温度稳定性好、机械品质因数较高和易烧结等特点, 在高温和高频领域有广阔的应用前景, 而且在自发极化翻转时不存在疲劳效应, 是用于非易失性铁电存储器 (FeRAM) 的理想材料[ 2, 3, 4]。此外, 铋层状共生结构化合物由于具有丰富的结构和物理性质, 在基础研究领域也备受关注。
本工作利用第一性原理计算的方法研究 iBTN的热力学稳定性和电子结构, 为其共生机制的探讨以及电子结构对电学性质的影响提供理论依据。
材料研究的传统方法试验周期长, 结果随机性大, 需要投入大量人力物力。近十几年来兴起的第一性原理计算研究不仅避免了上述缺点, 而且可设计性强, 同时可以从理论上对材料的性质做出合理解释, 为人们的研究提供明确指导。
所有相关的结构都采用几何优化得到其稳定结构, 同时计算出它们的总能。表1给出了Bi4Ti3O12的优化结果和文献参考值。原始结构所采用的晶胞参数全部来自于文献报告的结果[ 8, 9, 10, 11, 12, 13]。 iBTN 采用正交结构(I 2cm)[ 8], 如图1所示。Bi2O3有四种相结构(α、β、δ和γ), 优化采用最稳定的α相结构[ 9]。类似的, TiO2有三种晶型(金红石、锐钛矿和板钛矿), 这里采用金红石结构[ 12]。Nb2O5采用T-Nb2O5结构[ 13]。
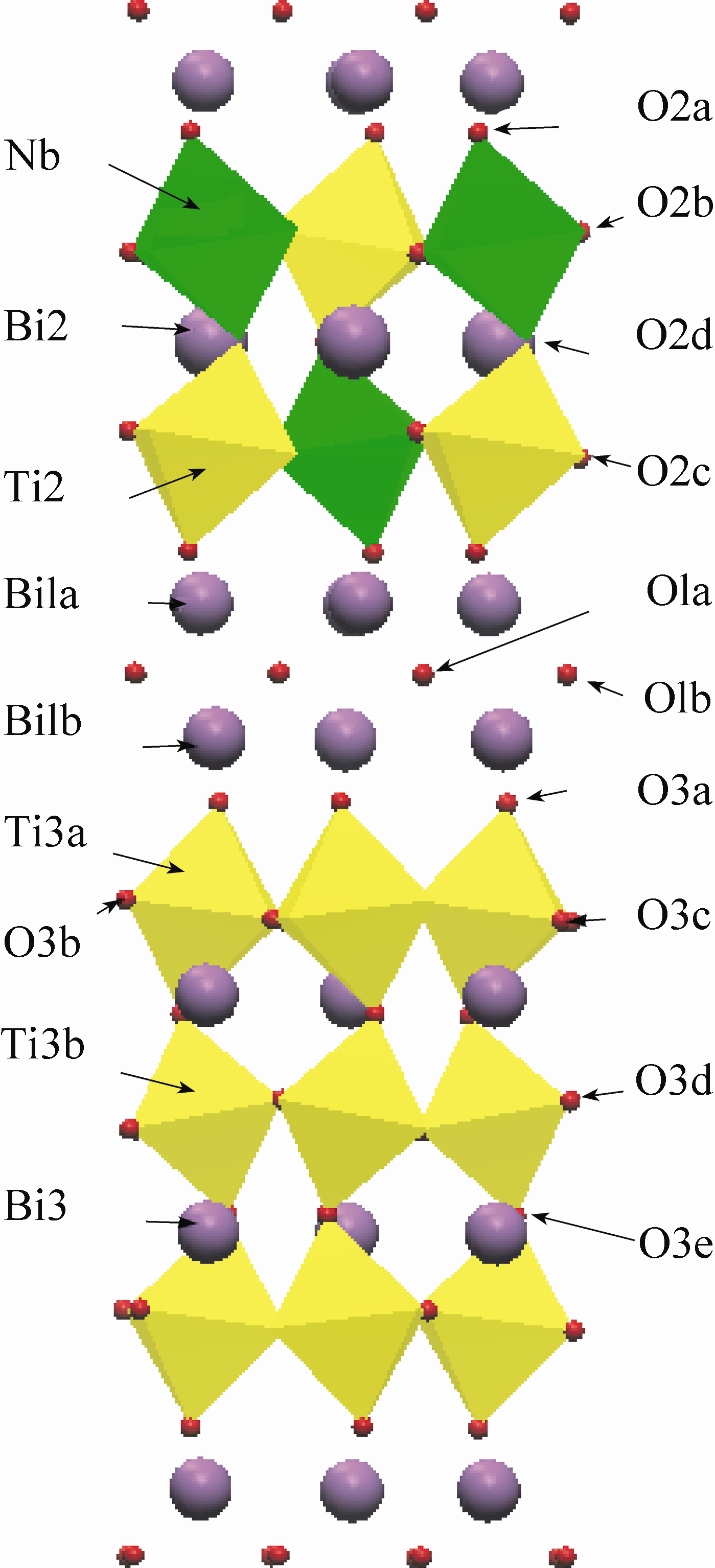 | 图1 iBTN的原子位置示意图Fig. 1 The orthorhombic structure of iBTN along [100] |
| 表1 优化得到Bi4Ti3O12晶格参数与文献的对比 Table 1 The calculated parameters of Bi4Ti3O12 and comparison with published literatures |
在合成Bi7Ti4NbO21的过程中, 避免Bi2O3、TiO2和Nb2O5的形成是非常重要的。为了根据原子化学势确定合适的合成条件, 我们基于不同化合物的总能(每个化学式单元)和组成元素的总能(每个原子)计算了它们的生成焓[ 14]。生成焓的计算依据由方程式(1~6)给出:
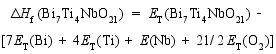 | (1) |
 | (2) |
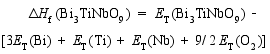 | (3) |
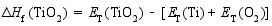 | (4) |
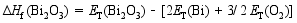 | (5) |
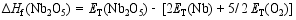 | (6) |
生成焓的计算使用的是在0 K下的总能, 假设随着温度的变化, 其值不会发生大的改变。
由于氧分子的总能不能按照周期性的结构计算得到, 因此我们根据热力学生成焓间接计算氧分子的总能。表2是氧分子总能的结果, 根据不同氧化物计算出值的偏差小于0.1%, BiT、BTN和 iBTN的生成焓所采用的氧分子总能基于三者的平均值。表3列出了 iBTN和其他相关化合物的生成焓数据。共生结构铁电体 iBTN相对于其组成单元BiT或BTN, 生成焓更小, 处在热力学相对稳定的状态, 但和同组成的BiT和BTN相比,处在较高的位置。因此, 共生结构 iBTN在热力学上处在亚稳态, 同组成的BiT和BTN相更为稳定, 这解释了实验上随着温度的升高, 首先生成BiT和BTN, 然后在较高温度下生成共生结构的现象[ 19]。
| 表2 采用不同氧化物计算得到的氧分子总能 Table 2 Calculated energy of oxygen with different oxides |
| 表3 计算得到的一些化合物的生成焓 Table 3 Calculated energy of oxygen with different oxides |
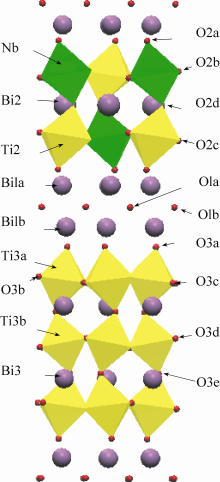
图2显示了结构优化后的 iBTN、BTN和BiT三种结构的态密度。 iBTN的带隙为0.796 eV, 其组成单元BTN和BiT的带隙为2.535和2.436 eV。共生结构铁电体 iBTN的带隙小于其组成单元BiT和BTN, 这主要是由于导带下移所造成的。
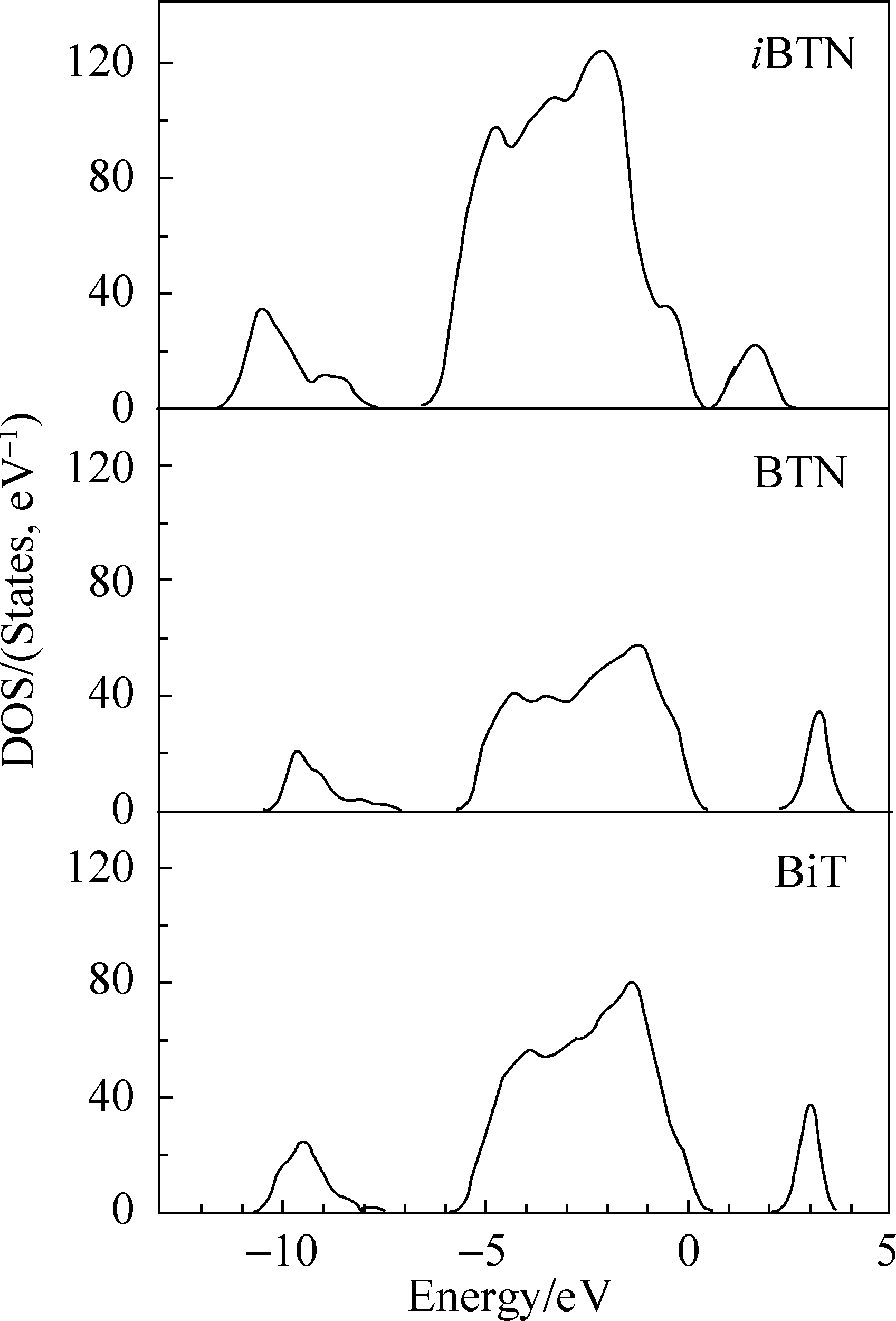 | 图2 iBTN、BTN和BiT的态密度Fig. 2 Total DOS of iBTN, BTN and BiT |
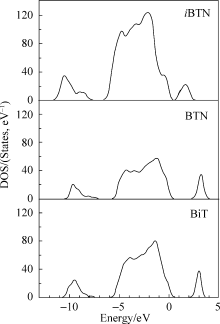
图3给出了共生结构铁电体 iBTN的态密度和不同元素的分态密度。价带由O2p、Ti5d、Nb5d、Bi6s和Bi6p态所组成, 其中O的2p态起主导作用; 导带主要是由于Ti和Bi的作用, 其中Ti起大部分作用, 而[TiO4]是钙钛矿层的主要组成单元, 说明钙钛矿层对导带的影响可能是决定性的。
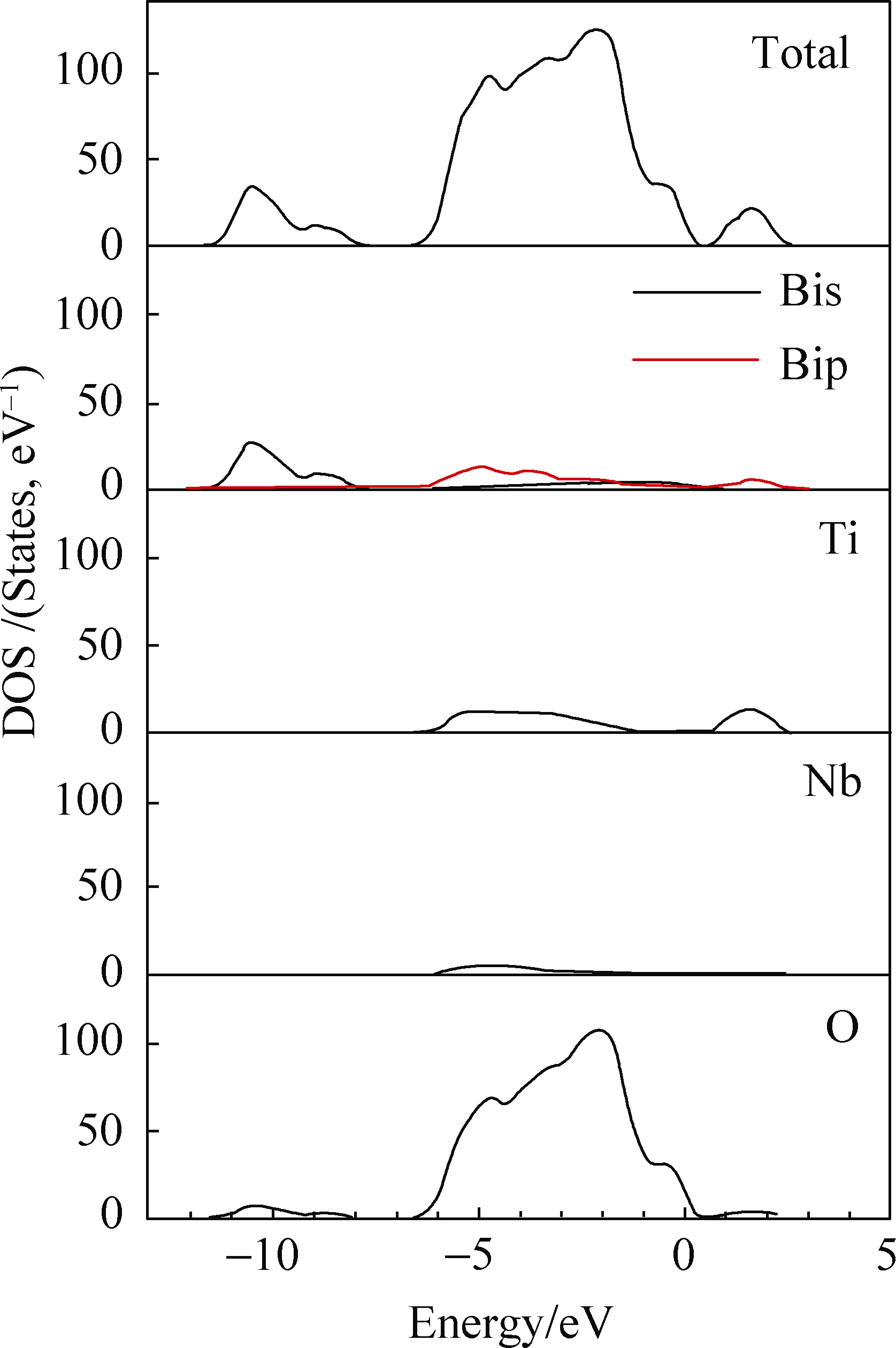 | 图3 iBTN的态密度和不同元素的分态密度Fig. 3 DOS and partial DOS for different elements of iBTN |
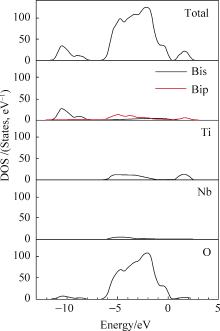
为了进一步分析态密度的构成, 对 iBTN中不同的原子作了分态密度图, 如图4所示。
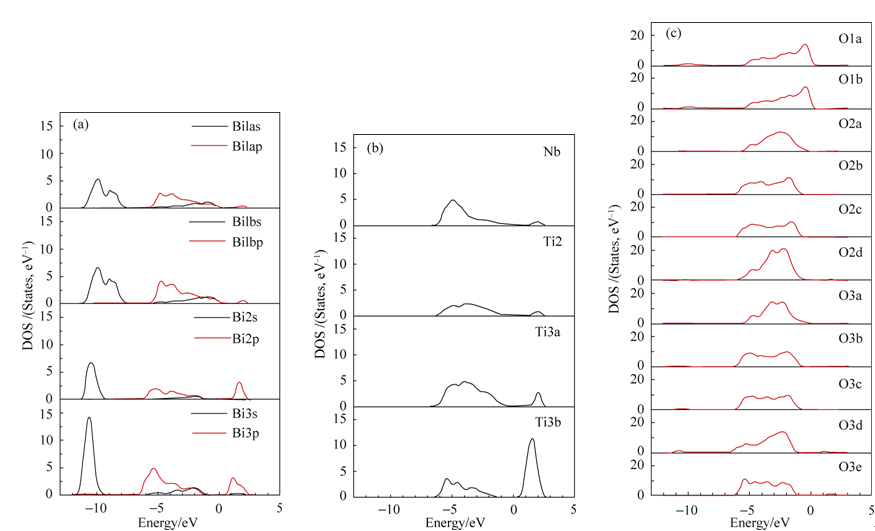 | 图4 iBTN中Bi原子(a), Ti原子和Nb(b), 以及各O原子(c)的分态密度Fig. 4 Partial DOS for Bi (a), Ti and Nb (b) and O (c) of Bi7Ti4NbO21 |
图4(a)是不同Bi原子的分态密度图, 在Bi原子中, 两层钙钛矿层中Bi2原子和三层钙钛矿层中Bi3原子对导带影响占主导, 这再次确定了钙钛矿层是影响导带的主要因素, 而铋氧层中的Bi影响极其有限。在导带的主要贡献者Ti原子中, 三层钙钛矿中间层的Ti原子(Ti3b)的影响最大, 这预示着电子在传输过程中会有优先选择的通道, 如图4(b)。钙钛矿中Bi原子(Bi2和Bi3)导带底相对于铋氧层中Bi限。在导带的主要贡献者Ti原子中, 三层钙钛矿中间层的Ti原子(Ti3b)的影响最大, 这预示着电子在传输过程中会有优先选择的通道, 如图4(b)。钙钛矿中Bi原子(Bi2和Bi3)导带底相对于铋氧气层中Bi原子处于较低的位置, 如图4(a); 钙钛矿层各中心原子(Ti和Nb)也共同参与减小了共生结构的带隙, 见图4(b)。图4(c)给出了氧原子的分态密度图, 二层钙钛矿中O2(a、b、c和d)和三层中O3(a、b、c、d和e)的价带顶比铋氧层中的氧原子O1(1和2)能量位置更低, 事实上, 这和导带底的趋势是一致的, 这再次确定了钙钛矿层同样也是造成共生结构相对于其组成单元带隙减小的影响因素。
运用第一性原理计算研究了铋层状共生结构铁电体Bi7Ti4NbO21及其相关化合物, 计算得到了它们的生成焓和态密度, 并对热力学稳定性、电子电导和带隙与结构的相互关系进行了阐述, 得到以下结论:
1) 共生结构铁电体 iBTN相对于同组成的BTN和BiT在热力学上处于亚稳状态, 这与文献结果是一致的;
2) 电子结构的计算结果表明, 钙钛矿层是材料电子电导的主要影响因素, 铋氧层几乎没有影响;
3) 共生结构 iBTN的带隙比组成单元都要小, 这个结果虽然有待实验验证, 但深入的计算发现, 钙钛矿层造成了导带底的下移, 从而导致带隙的减小。
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|