

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
掺锶量对羟基磷灰石形貌及其荧光特性的影响
[赵欣 , 王德平, 邱雯青, 叶松]
, 王德平, 邱雯青, 叶松]
, 王德平, 邱雯青, 叶松]
|
|


赵 欣(1988-), 女, 硕士研究生. E-mail:zhaoxin_cynthia@163.com
采用水热合成法制备了一系列具有不同微观形貌和荧光性能的掺锶羟基磷灰石粉末。通过X射线衍射、红外光谱、电子能谱、扫描电镜和荧光光谱表征样品的物相、形貌及荧光性能。结果表明: 所制备的样品形貌为1~ 3 μm的由微小晶粒聚集而成的球形颗粒, 但随着掺锶量的变化, 组成球形颗粒的微小晶粒形貌会出现较大的差异。 未掺锶羟基磷灰石晶粒形貌为短棒状, 随着掺锶量的增加, 微小晶粒逐渐变为片状而后转变为长棒状。样品能在紫外光(波长351 nm)激发下发出明亮的蓝色荧光(波长375~500 nm, 最强峰位432 nm), 且荧光强度随掺锶量增加先增强, 而后减弱, 在掺锶量为30mol%时达到最大。
Strontium Hydroxyapatite (Sr
羟基磷灰石(Ca10(PO4)6(OH)2, HAp)是脊椎动物骨骼中的重要矿物, 合成的HAp由于具有良好的生物相容性、对人体组织无毒性及良好的选择性吸附等特点而被广泛应用于骨组织修复[ 1]、药物载体[ 2]、色谱[ 3]或环境治理[ 4]等领域。另外, 当HAp的Ca2+被Sr2+等离子部分或全部取代后, 还能形成不同类型的固溶体, 并能促进成骨细胞增殖或抑制破骨细胞活性等[ 5]。
近年来, 有关HAp材料的多功能化的研究也受到了越来越多的关注, 并成为无机生物材料研究领域中的热点之一。例如, 可赋予HAp磁性能, 使其兼具良好的吸附性能和磁性能, 用于污水处理中重金属物质的分离[ 6], 或用作药物载体中的磁靶向定向给药[ 7]等。还可赋予HAp荧光性能, 用于药物缓释量的标定、药物缓释的监控及药物示踪等生物医学领域[ 8]。但是相关研究使用的都是容易光致褪色和易分解的荧光染料[ 9]、本身含有重金属离子的稀土元素[ 10]或半导体量子点材料[ 11]等, 影响材料的生物安全性。为改善这些亟需解决的问题, 本工作尝试将锶离子作为掺杂元素添加于HAp中, 并通过晶格缺陷作用, 形成一种荧光强度可调的HAp材料。重点考察掺锶量对HAp晶体形貌特征和荧光性能的影响, 并对荧光性能随锶离子含量的变化规律及其原理进行简要分析。
按化学计量比将Ca(NO3)2·4H2O和(NH4)2HPO4以及适量的柠檬酸三钠和模板剂CTAB溶于50 mL去离子水中, 剧烈搅拌30 min后, 将混合液移至高压釜中, 在180℃下保温24 h。冷却至室温后, 将溶液中的沉淀离心分离, 使用去离子水和乙醇反复清洗。而后在80℃红外干燥箱内干燥12 h, 得到最终的HAp粉末样品。
为了获得含锶HAp(SrHAp)样品, 用按摩尔比配制的Ca(NO3)2·4H2O和Sr(NO3)2混合物替换, 表1为不同掺锶量HAp配方标识。
| 表1 掺锶羟基磷灰石样品配方标识 Table 1 Formula of strontium hydroxyapatite samples |
使用D/max2550VB3+/PC型X射线粉末多晶衍射仪(XRD)对样品晶相分析; 使用EQUINOXSS/ HYPERION2000型红外光谱仪(FT-IR)对样品化学组成进行分析; 使用PHI 5000C ESCA System型光电子能谱(XPS)对样品中各离子的结合能进行分析; 使用970CRT型荧光分光光度计记录样品的荧光激发光谱和荧光发射光谱; 使用Quanta 200 FEG型场发射环境扫描电镜(SEM)对样品形貌进行观察。
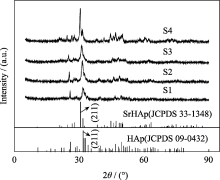
图1为不同样品的XRD图谱, 由图可知不含锶的HAp的XRD图谱与HAp (JCPDS 09-0432)对应, 而全锶HAp的XRD图谱则与SrHAp(JCPDS 33-1348)对应较好, 且峰型较为尖锐, 这表明锶的添加对样品的结晶性能影响较大。表2为样品S1、S2、S3 和 S4对应晶相、(211)晶面峰位及晶胞参数。由表2可知, 随着含锶量的增加, SrHAp(211)晶面衍射峰位向小角度方向偏移, 且偏移量随掺锶量的增大而增大。由于锶离子半径大于钙离子半径(Ca2+离子半径=0.100 nm; Sr2+离子半径=0.118 nm), 掺锶后会引起晶格畸变, 并使晶胞参数对应增大。
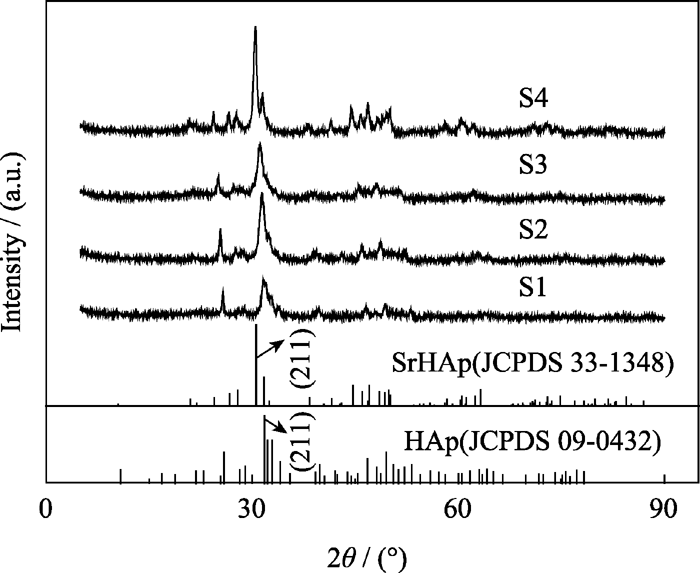 | 图1 样品S1、S2、S3和S4的XRD图谱Fig. 1 XRD patterns of samples S1, S2, S3 and S4 |
| 表2 不同样品对应晶相、(211)晶面峰位及晶胞参数 Table 2 Crystalline phase, peak position (211) and lattice parameters of different samples |
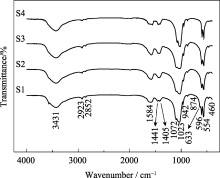
图2为不同样品的FTIR图谱。554、596 cm-1处是PO43-的弯曲振动峰, 942、1023、1072 cm-1处是PO43-的不对称伸缩振动峰, 874 cm-1处是HPO42-的振动峰, 这些基团表明了存在羟基磷灰石结构[ 12, 13], 与XRD的分析结果相符合; 1405、1441 cm-1处是CO32-的振动峰, 由制备过程水溶解空气中CO2产生[ 14]; 2852、2923 cm-1处是-CH2的振动峰, 为残留的表面活性剂CTAB产生; 3431 cm-1处振动带是羟基的特征振动峰。可见不同样品都存在PO43-、HPO42-、CO32-、H2O及有机碳链, 且不同掺锶量羟基磷灰石FTIR图谱没有明显变化, 表明其基团组成相同。
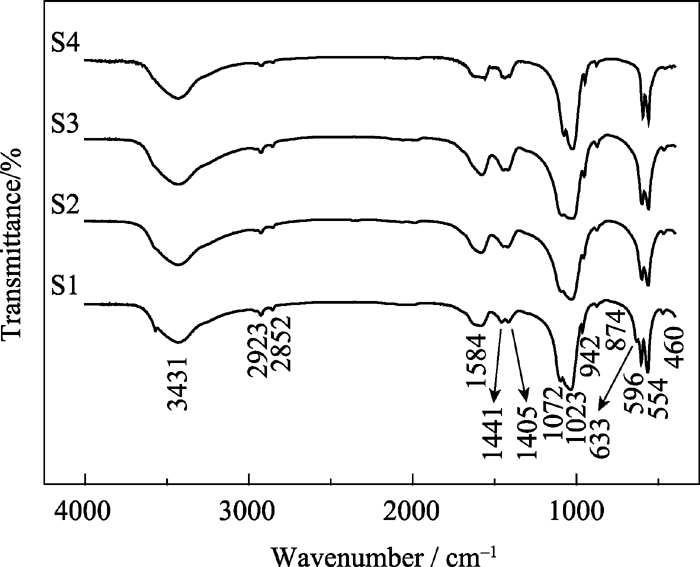 | 图2 样品S1、S2、S3和S4的FT-IR图谱Fig. 2 FTIR spectra of samples S1, S2, S3 and S4 |
图3为不同样品的SEM照片。由图可知不同掺锶量HAp颗粒呈球形均由微小晶粒聚集而成, 但随着掺锶量的不同, 组成球形颗粒的晶粒形貌发生了较大变化: 不掺锶量HAp组成颗粒的晶粒形貌为纳米短棒状结构(图3(a)), 随着掺锶量的增加, 当 Xsr为0.3时转变为片状结构(图3(b)), 当 Xsr增加至0.5时转变为棒状结构(图3(c)), 至 Xsr为1时则可以很明显观察到长棒状结构(图3(d))。
 | 图3 样品S1 (a)、S2 (b)、S3(c)和S4 (d)的SEM照片Fig. 3 SEM images of samples (a) S1, (b) S2, (c) S3 and (d) S4 |
SrHAp的形成包括成核和生长两个过程。在水热反应的前期, 由于HAp的溶度积很小, 因此成核速率高, 形成大量晶核。随着时间的延长晶体会发生取向生长。根据Zhong等[ 15, 16]提出的“负离子配位多面体生长基元模型”, 在 a轴方向生长基元主要是OH-Ca6, 带正电荷; 在 c轴方向生长基元主要是Ca-P6O24, 带负电荷。Ca2+在 c轴方向与生长基元的连接数量多于 a轴方向, 因此 c轴方向的生长速率高于 a轴方向, 导致通常形成棒状HAp。当未掺锶时, HAp的成核速率大于晶粒长大速度, 形成大量的纳米短棒状微小晶粒, 团聚形成的颗粒也较小(图3(a))。但当掺入锶后, Christoffersen等[ 17]认为锶对HAp晶体的生长有抑制作用, 张锐[ 18]实验证明锶的掺入对 c轴生长的抑制作用大于 a、 b轴, 因此随着掺锶量的增加, c轴的生长速率减小, 且接近或小于 a、 b轴的生长速率, 从而导致晶体形貌的长径比减小甚至出现片状结构。Christoffersen等[ 17]通过实验还证明锶对HAp的抑制作用随溶液中锶离子浓度增大而减弱。如图3所示, 当掺锶量增大到一定程度时, HAp晶体的形貌变为棒状且随掺锶量增加长径比增大。沉淀颗粒在生长过程中自组装形成球状颗粒的行为是晶粒团聚成球形颗粒长大时其表面能最小, 球状结构最稳定。
图4(a)和(b)分别为样品S1、S2、S3和S4的PL激发光谱和PL发射光谱。激发光在波长325~ 375 nm的紫外光范围内, 最大峰值为351 nm。各样品的激发光波长相同, 说明其发光机理相同。各样品的荧光发射波长范围都位于375~500 nm, 最大峰值为432 nm, 呈明亮的蓝色荧光。但随着掺锶量的改变, 各样品的荧光发射强度有较大变化。 Xsr由0增加到0.3时, 荧光强度随掺锶量的增加而增强, 且在 Xsr为0.3时达到最大值; Xsr由0.3增加到1时, 荧光强度随掺锶量的增加反而减弱, 全锶HAp荧光强度小于不掺锶的HAp。
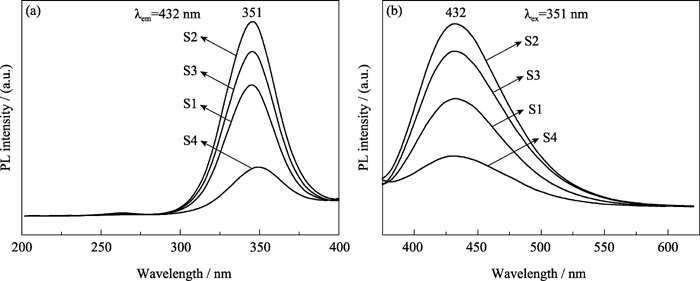 | 图4 样品S1、S2、S3和S4的PL激发光谱(a)和PL发射光谱(b)Fig. 4 Excitation (a) and emission (b) spectra of samples S1, S2, S3 and S4 |
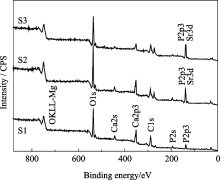
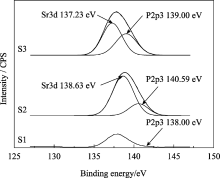
图5为样品S1、S2和S3的XPS全谱, 各峰位电子结合能列于表3中。由图5可以看出, 随着 Xsr从0、0.3到0.5的增加, Ca2p3的峰强逐渐减弱, 表明HAp晶体内Ca含量的逐渐减少, 晶格内的钙被取代; 同时 Xsr为0.3和0.5的HAp XPS谱中P2p3的峰比未掺锶的HAp XPS谱中P2p3的峰强度明显增大。这是由于锶Sr3d的峰与P2p3峰位接近, 相互重叠加强所致。图6为 Xsr为0.3和0.5的P2p3峰的分峰结果, 分别得到的P2p3和Sr3d的吸收峰表明锶进入了HAp晶格, 取代了钙的位置。这与XRD分析结果相符。
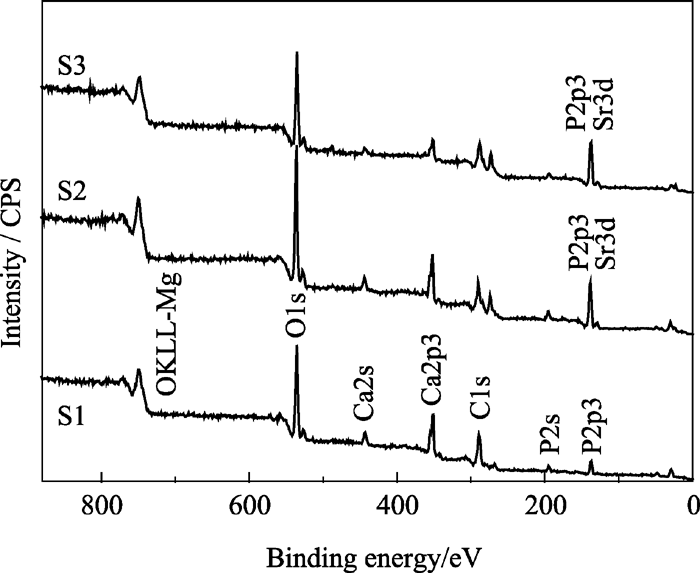 | 图5 S1、S2和S3样品的XPS全谱图Fig. 5 XPS spectra of samples S1, S2 and S3 |
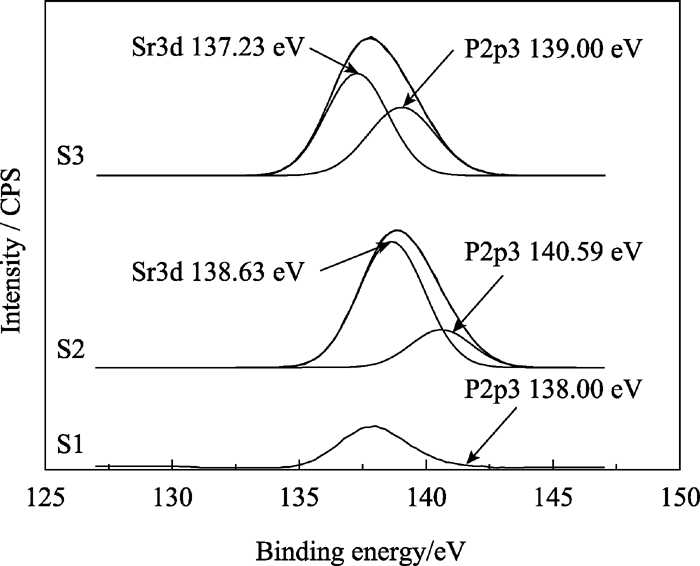 | 图6 样品S1、S2和S3在P2p3处的XPS分峰谱图Fig. 6 XPS deconvulation for P2p3 peaks of samples S1, S2 and S3 |
由表3各峰位的电子结合能数据分析可知, Xsr从0增加到0.3时, 晶格离子(O2-、Ca2+、P4+)的电子结合能都增大, 这表明晶格离子周围电子云密度减小。Zhang等[ 19]实验证明水热反应过程中存在的柠檬酸根分解产生的二氧化碳自由基(CO2·-)会进入HAp晶格间隙中并形成荧光发光中心。因而周围电子云密度的减小有利于CO2·-发光中心电子吸收和发射, 引起荧光强度的增大; 反之, Xsr从0.3增加到0.5时, 晶格离子的电子结合能减小, 电子云密度增大, 从而阻碍了CO2·-发光中心的电子吸收和发射, 引起荧光强度的减小。
| 表3 样品S1、S2和S3的各元素的电子结合能 Table 3 XPS peak energies of samples S1, S2 and S3 |
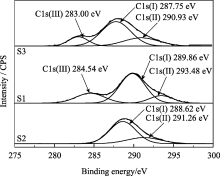
图7为对C进行分峰的分析结果。未掺锶的C1s具有两种成分: 288.62 eV的C-O键中C的电子结合能, 对照FTIR图谱分析结果可知, 它与CO32-有关, 也可能与对HAp荧光性能具有重要影响的CO2·-有关; 291.26 eV的C-H键中C的结合能, 对照FTIR图谱分析结果可知, 它与残留的CTAB中的-CH2基团有关。从图7还可以看到 Xsr为0.3和0.5的C电子结合能状态较未掺锶的样品而言, 分别多出了284.54和283.00 eV的峰位, 表明掺锶后C的电子状态发生了改变。这是因为锶离子半径大于钙离子半径使得锶的外层电子比钙的外层电子活跃, 进而在取代钙离子后, 会改变周围C的电子状态。这一新出现的C的状态有利于提高锶SrHAp中的CO2·-发光中心的电子吸收和发射比例, 使得掺锶后的SrHAp荧光强度增强。
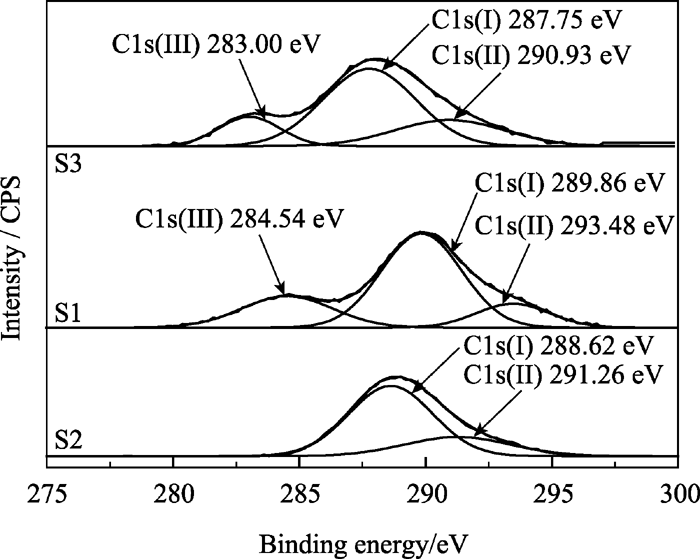 | 图7 样品S1、S2和S3在C1s处的XPS分峰谱Fig. 7 XPS deconvulation for C1s of samples S1, S2 and S3 |
根据荧光性能测试结果分析, 掺锶导致HAp中晶格离子结合能的变化对荧光性能的影响应该为主要因素, 而掺锶导致的CO2·-发光中心中C的电子结合能状态的变化对荧光性能的影响则为次要因素。由结果分析可知, 当掺锶量较低时, 上述两种效应将起叠加效应, 即荧光强度随着掺锶量的增加而增强, 且在 Xsr为0.3处达到最大值。但当掺锶量继续增多后, HAp的晶面间距持续增大, 从而导致晶格离子的结合能反而减少, 其结果是当 Xsr从0.3增加到1的过程中荧光强度的持续减少, 这与实验结果相一致。
采用水热合成法制备了 n(Sr)/ n(Sr+Ca)为0、0.3、0.5和1的SrHAp球状粉末, 样品能在紫外光的作用下(波长344 nm)发出明亮的蓝色荧光(波长350~570 nm, 最强峰位427 nm)。实验结果表明, 随着 n(Sr)/ n(Sr+Ca)从0到1的增加, 组成微球的晶粒形貌也相应地由短棒状( n(Sr)/ n(Sr+Ca)=0)转变为片状( n(Sr)/ n(Sr+Ca)=0.3)、短棒状( n(Sr)/ n(Sr+Ca)=0.5)直至形成长棒状( n(Sr)/ n(Sr+Ca)=1)结构的变化。但HAp晶体的荧光强度的变化却呈随掺锶量增加逐渐增强, 并在 n(Sr)/ n(Sr+Ca)为0.3时达到最大, 而后随掺锶量增加逐渐减弱的趋势。其原因与掺锶后HAp母相中的晶格离子的结合能变化、间隙杂质基团CO2·-发光中心的电子吸收和发射能力的变化等有关。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|