{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
微波辅助水热法快速合成介孔磷酸镍
[李旭影 , 谭涓, 杨建华, 刘靖, 杨菲]
, 谭涓, 杨建华, 刘靖, 杨菲]
, 谭涓, 杨建华, 刘靖, 杨菲]
|
|


李旭影(1984-), 女, 硕士研究生. E-mail:L_xying933@163.com
采用微波辅助水热法快速合成了介孔磷酸镍(MW-NiPO-2), 详细研究了在微波水热条件下微波晶化温度、微波辐射时间、碱度及Ni/P摩尔比对MW-NiPO-2合成的影响, 优化了合成条件. 并利用ICP、FT-IR、TEM和N2吸附等手段对比研究了溶胶-凝胶法和微波水热法合成样品的结构组成、比表面和孔道特征. 研究结果表明: 在微波辅助水热条件下, 在较低温度下(60℃)即可形成MW-NiPO-2; 在100℃下, 0.25 h即有MW-NiPO-2生成. 在微波辐射1 h, 碱度(OH-/P)为2.0、原料Ni/P比为0.7条件下, 能够合成有序度较高的MW-NiPO-2. MW-NiPO-2和 SG-NiPO-2具有相似的骨架结构和组成, 但其纳米管簇结构特征不明显, 表现出类似虫孔结构的状态. 微波辅助水热法合成的MW-NiPO-2较溶胶-凝胶法合成样品具有更大的比表面和孔容, 分别为334.4 m2/g和0.318 cm3/g.
Mesostructured nickel phosphate (MW-NiPO-2) was rapidly synthesized in the presence of cationic surfactant by a microwave assisted hydrothermal process. The synthesis parameters, microwave irradiation temperature and time, OH-/P and Ni/P molar ratios of the precursor, were systematically investigated and optimized. The characterizations of the materials obtained by the two different procedures, Sol-Gel method and microwave assisted hydrothermal process, were carried out by ICP, FT-IR, TEM and N2 physisorption. The results show that MW-NiPO-2 can be obtained at relatively low temperature (60℃). When the microwave radiation temperature was 100℃, the mesostructured phase formed in an extremely short period about 0.25 h. Ordered MW-NiPO-2 can be obtained under microwave irradiation for 1 h at 100℃, when OH-/P and the Ni/P molar ratios of the precursor are 2.0 and 0.7, respectively. In contrast, it took at least 24 h for completion
自M41S系列硅基介孔材料被发现以来, 采用表面活性剂自组装法合成非硅基介孔材料, 特别是含过渡金属元素的介孔材料, 如氧化物、硫化物和磷酸盐等, 引起了广泛关注. 这类材料由于其特有的元素组成、高比表面积及较大的孔径, 在催化、吸附与分离、高分子材料合成等领域有着重要的应用前景, 有望为介孔材料开辟新的应用领域[ 1, 2, 3, 4]. 2008年, 本课题组首次采用溶胶-凝胶法合成了具有纳米管簇结构的新型介孔磷酸镍材料NiPO-1和NiPO-2[ 5], 其基本结构单元为中空的纳米管簇, NiPO-1结构中的纳米管单元较长, 而NiPO-2的则较短. 介孔磷酸镍在大分子烯烃的环氧化反应中表现出很高的活性和选择性, 且反应过程中无Ni2+的浸出[ 5], 特别是它们的孔道尺寸较大(约为VSB-5孔道尺寸的4倍), 可以催化大分子底物的反应, 因此具有良好的应用前景.
近年来, 将微波加热方式应用到无机多孔材料合成领域引起人们的广泛兴趣[ 6, 7, 8, 9, 10, 11, 12, 13]. 微波辐射加热使合成釜内部受热均匀、升温快, 避免了浓度和温度梯度的影响, 具有反应速度快、相选择性和形貌可控等特点[ 6, 14, 15, 16]. 在介孔材料合成领域, 采用该方法已成功合成了多种硅基(MCM-41[ 7, 8]、MCM-48[ 10]、SBA-15[ 11]、SBA-16[ 12]等)和非硅基(TiO2[ 13]、Nd2O3[ 14]、氧化镓[ 15]等)介孔材料. 本工作尝试采用微波辅助水热法合成介孔磷酸镍, 详细研究在微波辅助水热条件下MW- NiPO-2的合成规律, 并与在电加热条件下采用溶胶-凝胶法合成的样品进行了对比研究.
将3.64 g十六烷基三甲基溴化铵(CTAB, AR, 天津科密欧化学试剂有限公司)溶于15 mL去离子水中, 充分溶解后加入1.15 g H3PO4(85%, 沈阳化学试剂厂), 60℃酸化1 h, 然后采用25 % 四甲基氢氧化铵(TMAOH, AR, 北京金科美化工产品有限公司) 调节溶液的pH为8~10, 搅拌均匀, 再称取一定量的 Ni(NO3)2·6H2O(AR, 天津市科密欧化学试剂有限公司)溶解于15 mL去离子水中, 加入到上 述溶液, 此时合成体系中物料的摩尔配比为: 1.0H3PO4:(0.3~0.9)Ni(NO3)2·6H2O:1.0CTAB:(1.9~2.3)TMAOH: 200H2O, 搅拌30 min后, 放入微波消解仪(MDS-6型自动变频温压双控微波消解/萃取仪)中, 在40~140℃下微波辐射0.25~2 h, 产品经过滤、洗涤、干燥, 得到原粉样品, 记为MW-NiPO-2; 在电加热条件下采用溶胶-凝胶法合成的样品[ 16], 记为SG-NiPO-2.
原粉样品加入到0.10 mol/L的萃取剂CH3COONa /EtOH中, 在78℃下萃取2次, 得到介孔MW-NiPO-2 /SG-NiPO-2样品.
定义在微波辐射条件为100℃, 1 h, 原料中OH-/P为2.0、Ni/P为0.7的条件下合成样品的[100]衍射峰强度为 I0, 其它样品的 [100]衍射峰的强度为 Is, Is/ I0 即为相对强度.
XRD采用日本理学D/MAX-2400型X射线衍射仪测定, 辐射源为Cu-Kα (λ=0.154 nm), 管电压为 40 kV, 管电流为100 mA. TEM采用Tecnai G220 S-Twin透射电子显微镜, 加速电压为300 kV. FT-IR光谱表征在Bruker公司的TENSOR27 型红外光谱仪上完成.
ICP采用Therm Jarrell Ash IRIS/AP测定样品中Ni与P的含量. 样品溶于一定浓度的HCl溶液中, 定容后测试. N2吸附等温线采用Micrometrics 2010物理吸附仪, 样品的比表面积通过BET方法求得, 孔容及孔径分布由BJH方法计算.
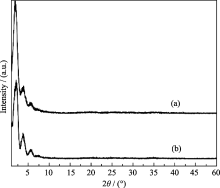
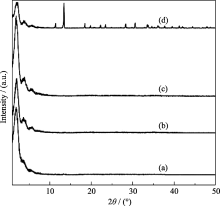
实验采用微波辅助水热合成法, 在原料中OH-/P比为2.0, Ni/P比为0.7, 100℃下微波辐射1 h条件下合成了介孔磷酸镍MW-NiPO-2, 其XRD结果示于图1中. 由图1(a)可见, MW-NiPO-2样品在小角范围内出现z峰[ 5, 16]. 此外, 其图谱在11.33°、13.33°和18.40°等高角处未出现致密的磷酸镍晶相产物的衍射峰[ 17], 说明在微波辅助水热条件下可以快速合成纯相的介孔磷酸镍MW-NiPO-2.
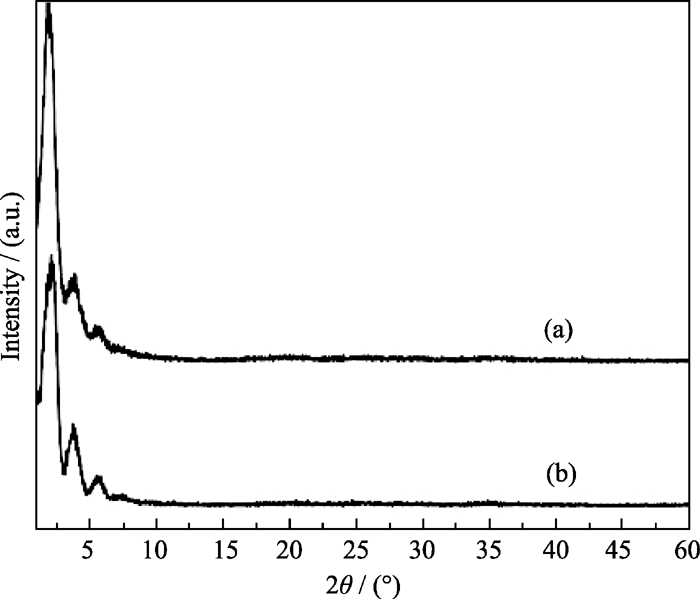 | 图1 MW-NiPO-2(a)和SG-NiPO-2(b)样品的XRD图谱Fig. 1 XRD patterns of as-synthesized MW-NiPO-2 (a) and SG-NiPO-2 (b) samplesConditions: OH-/P=2.0, Ni/P=0.7, 100℃/1 h |
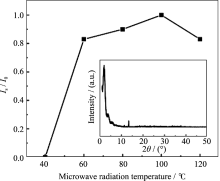
2.2.1 微波辐射温度的影响
在微波辐射时间为1 h、OH-/P为2.0, 原料Ni/P为0.7的条件下, 研究了不同辐射温度对合成MW- NiPO-2的影响, 样品XRD图谱中介孔相产物[100]衍射峰的相对强度变化示于图2. 由图2可知, 当微波辐射温度为40℃时, 样品中无介孔相产物生成, 主要为无定型物; 升高温度至60℃, 开始出现介孔相产物MW-NiPO-2的衍射峰, 说明在微波水热条件下, 较低温度下(60℃)即可生成MW-NiPO-2. 这可能是由于物质分子偶极振动同微波振动具有相似的频率, 微波加热方式是从物质分子出发的[ 18], 不但使物料快速升温, 受热均匀, 同时也促进了介孔相的形成. 随着温度的升高, 产物衍射峰的相对强度逐渐增强; 当温度达100℃时, 样品[100]衍射峰的强度达到最高; 温度继续升高, MW-NiPO-2的衍射峰强度降低, 并在高角处出现致密的磷酸镍晶相产物的衍射峰[ 17](如图2插图), 说明介孔相产物逐渐减少, 并向致密相磷酸镍晶体转化. 因此, 微波辅助水热合成MW-NiPO-2的最佳晶化温度为100℃.
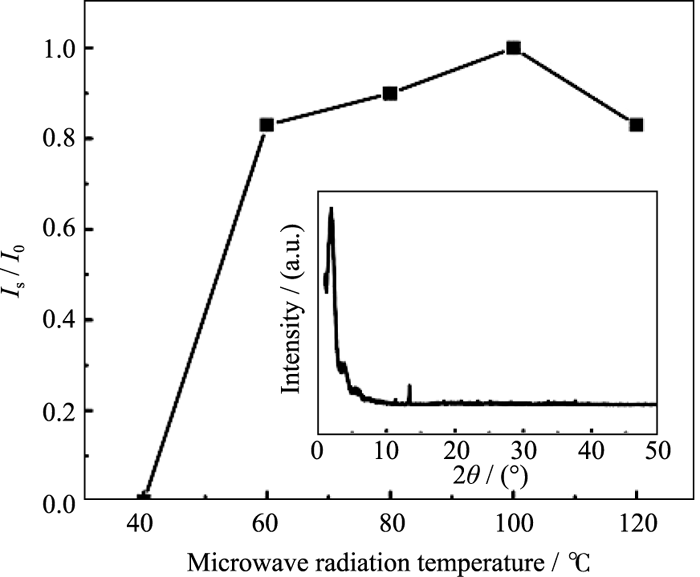 | 图2 微波辐射温度对合成样品[100]衍射峰相对强度的影响(插图为120℃时合成样品的XRD图谱)Fig. 2 Effect of radiation temperature on the evolution of Is/ I0 of MW-NiPO-2 (Inset is XRD pattern of sample synthesized at 120℃)Conditions: OH-/P=2.0, Ni/P=0.7, 1 h |
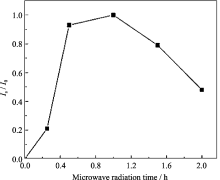
2.2.2 微波辐射时间的影响
在微波辐射温度为100℃、OH-/P为2.0, 原料Ni/P为0.7的条件下, 研究了不同微波辐射时间对合成MW-NiPO-2的影响, 样品的XRD图谱中[100]
衍射峰的相对强度如图3所示. 由图3可以看出, 当初始凝胶未进行微波辐射时, 样品为无定形物, 无介孔相产物生成; 当微波辐射时间为0.25 h时, 开始出现介孔相MW-NiPO-2的衍射峰, 说明微波水热条件促进了介孔相的快速形成. 这是由于微波与分子的耦合能力依赖于分子的性质, 从而有可能控制相选择性的可能[ 6], 对比SG-NiPO-2的合成规律[ 5]可以看出, 在微波水热条件下, 无机的镍离子和磷酸根在表面活性剂表面自组装生成介孔磷酸镍的时间非常短. 延长微波辐射时间至0.5 h, MW-NiPO-2的衍射峰相对强度迅速增大, 表明样品中MW-NiPO-2产物增加; 微波辐射时间延长至1 h时, 介孔相[100]衍射峰相对强度略有增大; 此后, 继续延长辐射时间, [100]衍射峰强度逐步降低, 磷酸镍晶体逐渐增多, 说明微波辐射时间超过1 h, MW-NiPO-2易向磷酸镍晶体转化. 因此, 最佳的微波辐射时间为1 h.
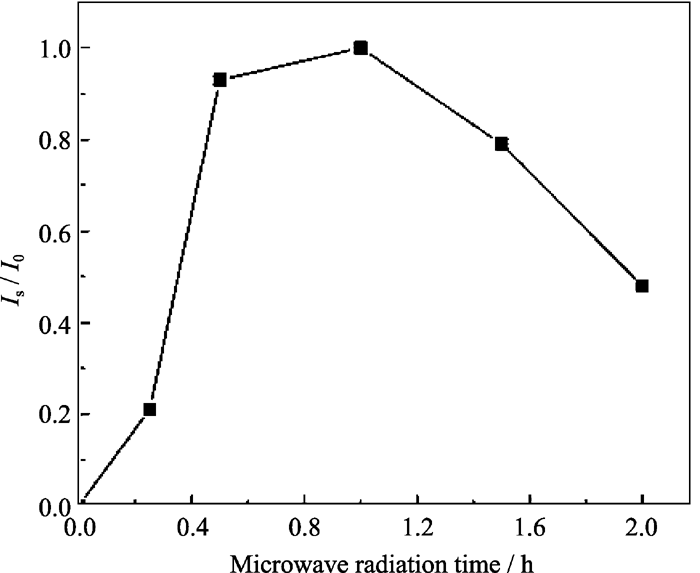 | 图3 微波辐射时间对合成样品[100]衍射峰相对强度的影响Fig. 3 Effect of radiation time on the evolution of Is/ I0 of MW-NiPO-2Conditions: OH-/P=2.0,Ni/P=0.7, 100℃ |
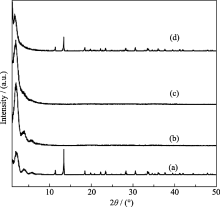
2.2.3 碱度对合成MW-NiPO-2的影响
在微波辐射温度为100℃, 时间为1 h, 原料中Ni/P摩尔比为0.7的条件下, 研究了初始凝胶中碱度(OH-/P)对合成MW-NiPO-2的影响, 其XRD图谱示于图4中. 由图4可知, 当初始凝胶的碱度(OH-/P) 为1.9时, 样品的XRD图谱中同时存在介孔相产物和致密相晶体的衍射峰, 说明合成产物为MW-NiPO-2和磷酸镍晶体的混合物; 增加OH-/P至2.0时, 晶体磷酸镍的衍射峰消失, 图谱中仅出现了较强MW-NiPO-2的衍射峰; 继续增加OH-/P至2.1时, 介孔相[100]衍射峰的相对强度变化不大, 而[200]和[300]衍射峰却几乎消失, 说明随着碱度的增大, MW-NiPO-2的结构有序度降低, 层垛状排列的纳米管簇转变为无序排列; 当OH-/P为2.3时, 谱图中介孔相的衍射峰明显降低, 并且有磷酸镍晶体的衍射峰出现. 这是由于体系中的pH值影响着Ni2+和PO43-在表面活性剂表面聚合的速度和状态, 因此OH-/P对MW-NiPO-2的合成影响较大, 较纯的介孔相产物出现的相区较窄, 仅在2.0~2.1之间, 这个范围之外都会导致磷酸镍晶体的产生.
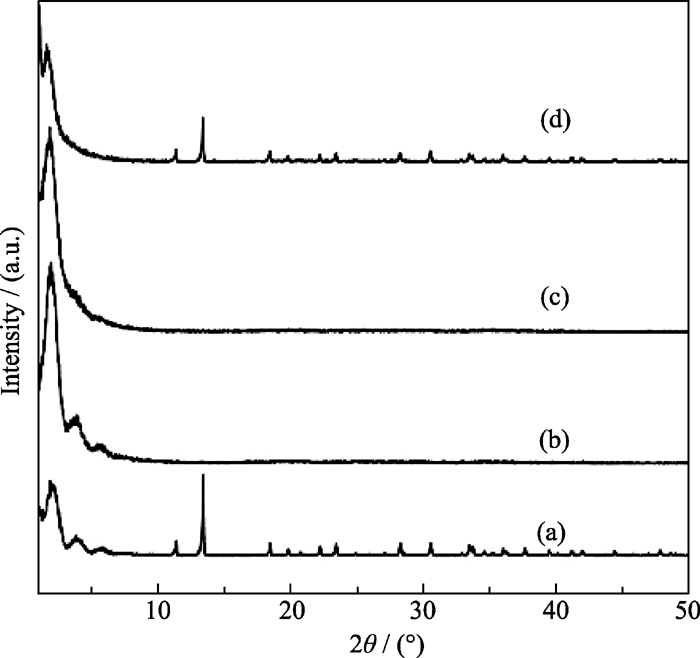 | 图4 不同碱度条件下合成MW-NiPO-2样品的XRD图谱Fig. 4 XRD patterns of as-synthesized MW-NiPO-2 samples with different OH-/P (a) 1.9; (b) 2.0; (c) 2.1; (d) 2.3Conditions: OH-/P=2.0,Ni/P=0.7, 100℃/1h |
2.2.4 原料Ni/P摩尔比的影响
在微波辐射温度为100℃, 时间为1 h, OH-/P为2.0条件下, 研究了初始凝胶中的Ni/P摩尔比对合成MW-NiPO-2的影响, 其XRD图谱如图5所示. 由图5可见, 当初始凝胶中Ni/P摩尔比为0.3时, 介孔相产物的[100]衍射峰较强, 而[200]和[300]衍射峰强度较弱, 说明样品结构有序度较低; 增加Ni/P比至0.5时, 谱图中介孔相的[100]衍射峰强度降低, 而[200]和[300]衍射峰明显增强, 说明MW-NiPO-2的结构有序度增强; 增加Ni/P比至0.7时, MW-NiPO-2的衍射峰强度达到最高; 继续提高Ni/P摩尔比, 合成样品中开始出现致密相晶体, 同时MW-NiPO-2产物减少. 因此, 合成MW-NiPO-2的最佳原料Ni/P比为0.7. 合成样品的ICP数据表明(表1), 初始凝胶中不同Ni/P比合成的MW-NiPO-2的骨架结构中实际Ni/P均为~1.6.
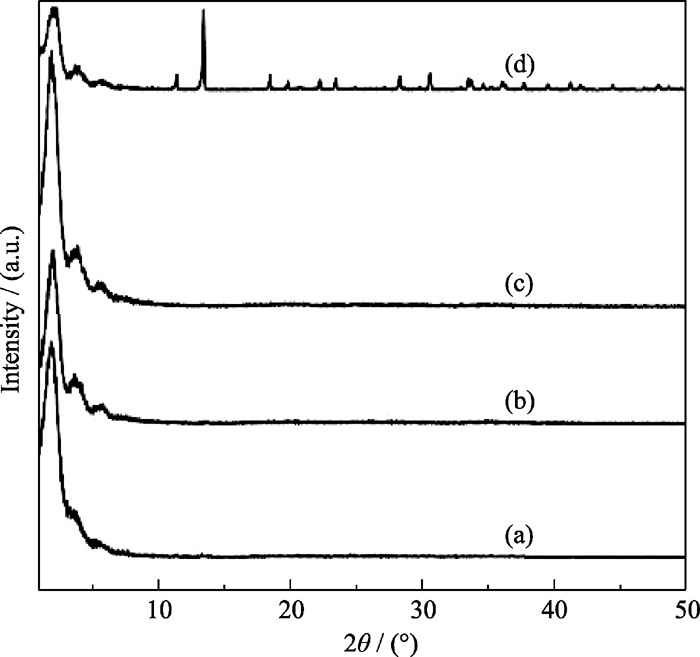 | 图5 不同原料Ni/P条件下合成MW-NiPO-2样品的XRD图谱Fig. 5 XRD patterns of MW-NiPO-2 samples synthesized with different Ni/P molar ratios (a) 0.3, (b) 0.5, (c) 0.7, (d) 0.9Conditions: OH-/P=2.0, Ni/P=0.7, 100℃/1 h |
| 表1 不同原料Ni/P合成的MW-NiPO-2样品的元素分析数值 Table 1 ICP data of MW-NiPO-2 samples synthesized with different Ni/P molar ratios in initial gel |
对在优化条件下合成的MW-NiPO-2样品进行了结构组成表征, 并与SG-NiPO-2样品进行了对比研究.
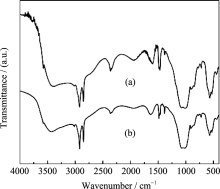
MW-NiPO-2的FT-IR图谱(图6)与 SG-NiPO-2相比差别不大, 说明二者的骨架组成特征基本相同. 在其谱图中, 2918, 2850, 1460, 1375, 以及720 cm-1处出现模板剂C16TMA+的特征吸收峰; 在1000~ 1100 cm-1之间出现两个较强的吸收峰为PO4的伸缩振动峰; 在400~700 cm-1范围的吸收峰为骨架中磷氧键及镍氧键的弯曲振动峰[ 5, 16]. 在3400 cm-1左右出现了很宽的羟基吸收峰, 表明其骨架结构中包含了大量的羟基, 这与介孔磷酸镍的亲水性特征及其较低聚合度的骨架相关[ 19].
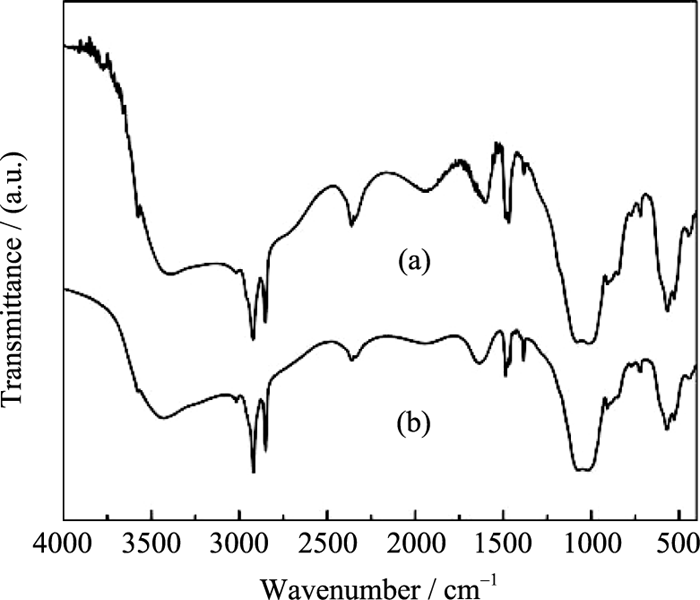 | 图6 MW-NiPO-2 (a)和SG-NiPO-2 (b) 样品的FT-IR图谱Fig. 6 FT-IR spectra of as-synthesized (a) MW-NiPO-2; (b) SG-NiPO-2 MW-Nipo-2 preparation condition OH-/P=2.0, Ni/P=0.7, 100℃/1 h |
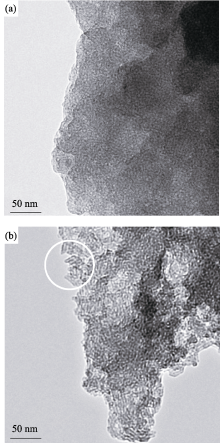
图7为MW-NiPO-2和SG-NiPO-2原粉样品的TEM照片. 由图7(b)可见, SG-NiPO-2在部分区域表现出中空的短纳米管(图中白圈部分), 大部分区域表现为虫孔结构. 虽然MW-NiPO-2的XRD图谱与SG-NiPO-2相似, 但是其纳米管簇结构特征更不明显, 表现出类似虫孔结构的状态. 影响其微观结构差别的原因可能是微波水热条件促进了介孔相的快速形成, 因此抑制了纳米管单元的生长长度.
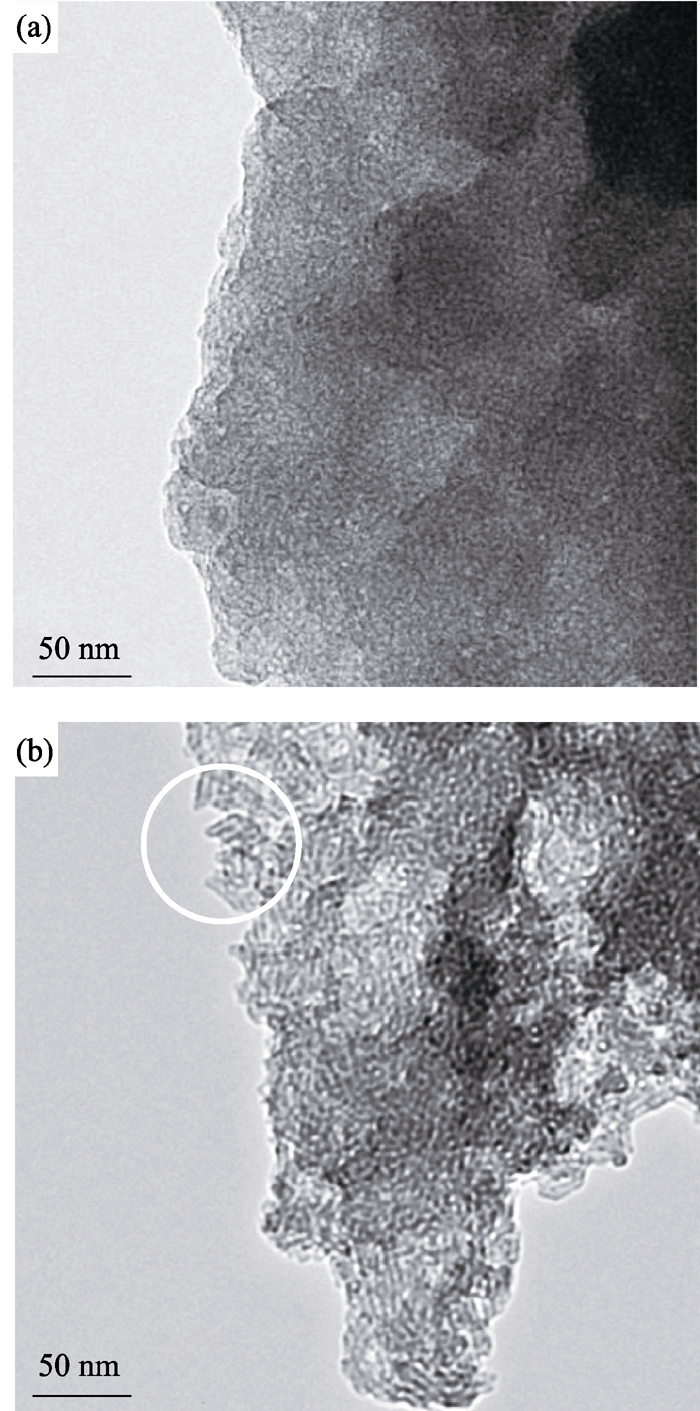 | 图7 MW-NiPO-2 (a)和 SG-NiPO-2 (b)样品的TEM照片Fig. 7 TEM images of as-synthesized mesoporous nickel phosphate samples(a) MW-NiPO-2; (b) SG-NiPO-2 |
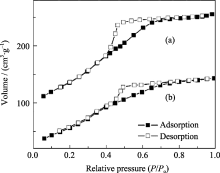
MW-NiPO-2和SG-NiPO-2样品的N2吸附-脱附曲线示于图8中. 由图可见, 这两种材料的吸附曲线都属于IV型, 是典型的介孔材料的吸附曲线. MW-NiPO-2表现出典型的H2型滞后环, 位于0.4~0.8相对压力范围内, 孔道结构为墨水瓶型, 与SG-NiPO-2的图形相似, 但迟滞环更大. SG- NiPO-2比表面和孔容分别为223.5和0.220 cm3/g, 而采用微波水热合成的MW-NiPO-2比表面和孔容明显增加, 分别达334.4和0.318 cm3/g.
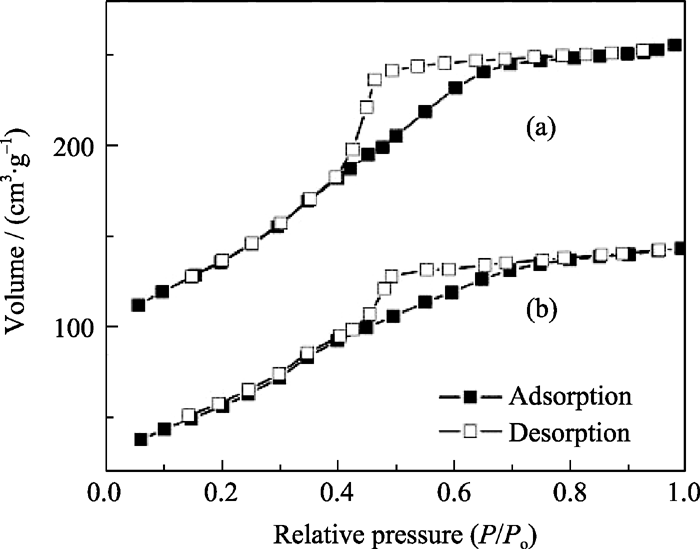 | 图8 MW-NiPO-2(a)和SG-NiPO-2(b)样品的吸附-脱附曲线Fig. 8 Adsorption-desorption isotherm of extracted samples (a) MW-NiPO-2; (b) SG-NiPO-2 |
采用微波辅助水热法可以快速合成介孔磷酸镍(MW-NiPO-2). 微波辐射温度、辐射时间、OH-/P及Ni/P摩尔比的选择是得到有序度较高的MW-NiPO-2的重要因素. 与溶胶-凝胶法相比, 生成介孔磷酸镍的温度较低(60℃), 并且时间很短(100℃下仅需0.25 h); OH-/P对MW-NiPO-2的合成影响很大, 纯相产物仅出现2.0~2.1之间. 当微波辐射温度为100℃, 辐射时间为1h, OH-/P摩尔比为2.0, Ni/P摩尔比为0.7时, 能够得到有序度较高的纯相MW-NiPO-2. ICP、FT-IR、TEM和N2吸附研究表明, MW-NiPO-2和 SG-NiPO-2具有相似的骨架结构和组成, 但其纳米管簇结构不明显, 表现出类似虫孔结构的状态. 此外, 微波水热法合成的MW- NiPO-2具有更大的比表面和孔容, 分别为334.4和0.318 cm3/g, 与溶胶-凝胶法合成的样品相比具有明显的优势.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|